
- DEPOD Home
- About DEPOD
- User Manual
- Human Phosphatases
- Protein Substrates
- Non-Protein Substrates
- Pathway Mapping
- Dephosphorylation Network
- ID Mapping
- Download
- Acknowledgement
- Contact
- Imprint
- Links
- Ensembl
- Entrez Gene
- HGNC
- QuickGO
- Gene Expression Atlas
- OMIM
- UniProt
- IntEnz
- BRENDA
- LocDB
- M-CSA
- InterPro
- Pfam
- CATH
- Gene3D
- PDB
- PDBe
- DrugPort
- ModBase
- SwissModel
- ChEMBL
- Phospho.ELM
- PhosphoSitePlus
- NetworKIN
- HPRD
- PSICQUIC
- STRING
- IntAct
- MINT
- KEGG Pathway
- Reactome Pathway
- Pathway Commons
- PubMed
- ArchSchema
- Cytoscape Web
- Europe PMC
- EKPD
- SwissLipids
 Statistics
Statistics
194 phosphatases have substrate data;
--------------------------------
336 protein substrates;
83 non-protein substrates;
1215 dephosphorylation interactions;
--------------------------------
299 KEGG pathways;
876 Reactome pathways;
--------------------------------
last scientific update:
11 Mar, 2019
Top
PTEN
| Gene Name | PTEN (QuickGO) | (A quick tutorial to explore the interctive visulaization) |
| Synonyms | PTEN, MMAC1, TEP1 | |
| Protein Name | PTEN |
|
| Alternative Name(s) |
Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase PTEN;3.1.3.16;3.1.3.48;3.1.3.67;Mutated in multiple advanced cancers 1;Phosphatase and tensin homolog; | |
| EntrezGene ID | 5728 (Comparitive Toxicogenomics) | |
| UniProt AC (Human) | P60484 (protein sequence) | |
| Enzyme Class | EC 3.1.3.16 3.1.3.48 3.1.3.67 (BRENDA | |
| Molecular Weight | 47166 Dalton | |
| Protein Length | 403 amino acids (AA) | |
| Genome Browsers | NCBI | ENSG00000171862 (Ensembl) | UCSC | |
| Crosslinking annotations | Query our ID-mapping table | |
| Orthologues | Quest For Orthologues (QFO) | GeneTree | |
| Classification | Superfamily: Class I Cys-based PTPs --> Family: DSP --> Subfamily: PTEN-like | Historic class: Class I Cys-based PTPs --> VH1-like or DSPs --> PTENs | CATH ID: 3.90.190.10 | SCOP Fold: CC1 | |
| Phosphatase activity | active | Catalytic signature motif: HCKAGKGR | |
| Phosphorylation Network | Visualize | |
| Domain organization, Expression, Diseases | (show / hide) |
| Protein Domain | InterPro | Pfam | SMART | 3D Structure | PDB | DrugPort | ModBase | SwissModel |
| Polypeptide organization (Pfam) | No Pfam domain found | ||
| Catalytic Site | Mechanism and Catalytic Site Atlas | ||
| Gene Expression | Gene Expression Atlas | Diseases | OMIM | DisGeNet | COSMIC | ClinVar |
| Protein-protein interactions | STRING | IntAct | MINT | BioGRID | ||
| Phosphosites | Phospho.ELM | PhosphoSitePlus | ||
| Localization, Function, Catalytic activity and Sequence | (show / hide) |
| Localization (UniProt annotation) | |||
| Cytoplasm NucleusNucleus, PML body Note=Monoubiquitinated form is nuclear Nonubiquitinated form iscytoplasmic Colocalized with PML and USP7 in PML nuclear bodies(PubMed:18716620) XIAP/BIRC4 promotes its nuclear localization(PubMed:19473982) Isoform alpha: SecretedNote=May be secreted via a classical signal peptide and reenterinto cells with the help of a poly-Arg motif | |||
| Function (UniProt annotation) | |||
| Tumor suppressor Acts as a dual-specificity proteinphosphatase, dephosphorylating tyrosine-, serine- and threonine-phosphorylated proteins Also acts as a lipid phosphatase,removing the phosphate in the D3 position of the inositol ringfrom phosphatidylinositol 3,4,5-trisphosphate,phosphatidylinositol 3,4-diphosphate, phosphatidylinositol 3-phosphate and inositol 1,3,4,5-tetrakisphosphate with order ofsubstrate preference in vitro PtdIns(3,4,5)P3 > PtdIns(3,4)P2 >PtdIns3P > Ins(1,3,4,5)P4 (PubMed:26504226) The lipid phosphataseactivity is critical for its tumor suppressor functionAntagonizes the PI3K-AKT/PKB signaling pathway bydephosphorylating phosphoinositides and thereby modulating cellcycle progression and cell survival The unphosphorylated formcooperates with AIP1 to suppress AKT1 activation Dephosphorylatestyrosine-phosphorylated focal adhesion kinase and inhibits cellmigration and integrin-mediated cell spreading and focal adhesionformation Plays a role as a key modulator of the AKT-mTORsignaling pathway controlling the tempo of the process of newbornneurons integration during adult neurogenesis, including correctneuron positioning, dendritic development and synapse formationMay be a negative regulator of insulin signaling and glucosemetabolism in adipose tissue The nuclear monoubiquitinated formpossesses greater apoptotic potential, whereas the cytoplasmicnonubiquitinated form induces less tumor suppressive ability Inmotile cells, suppresses the formation of lateral pseudopods andthereby promotes cell polarization and directed movement Isoform alpha: Functional kinase, like isoform 1 itantagonizes the PI3K-AKT/PKB signaling pathway Plays a role inmitochondrial energetic metabolism by promoting COX activity andATP production, via collaboration with isoform 1 in increasingprotein levels of PINK1 | |||
| Catalytic Activity (UniProt annotation) | |||
| Phosphatidylinositol 3,4,5-trisphosphate +H(2)O = phosphatidylinositol 4,5-bisphosphate + phosphate [a protein]-serine/threonine phosphate + H(2)O= [a protein]-serine/threonine + phosphate Protein tyrosine phosphate + H(2)O = proteintyrosine + phosphate | |||
| Protein Sequence | |||
| MTAIIKEIVSRNKRRYQEDGFDLDLTYIYPNIIAMGFPAERLEGVYRNNIDDVVRFLDSKHKNHYKIYNLCAERHYDTAK FNCRVAQYPFEDHNPPQLELIKPFCEDLDQWLSEDDNHVAAIHCKAGKGRTGVMICAYLLHRGKFLKAQEALDFYGEVRT RDKKGVTIPSQRRYVYYYSYLLKNHLDYRPVALLFHKMMFETIPMFSGGTCNPQFVVCQLKVKIYSSNSGPTRREDKFMY FEFPQPLPVCGDIKVEFFHKQNKMLKKDKMFHFWVNTFFIPGPEETSEKVENGSLCDQEIDSICSIERADNDKEYLVLTL TKNDLDKANKDKANRYFSPNFKVKLYFTKTVEEPSNPEASSSTSVTPDVSDNEPDHYRYSDTTDSDPENEPFDEDQHTQI TKV |
| Motif information from Eukaryotic Linear Motif atlas (ELM) | (show / hide) |
| ELM Motif | Motif Instance | Description | Motif Regex |
| CLV_C14_Caspase3-7 | 381-DTTDS-385 | Caspase cleavage motif | [DSTE][^P][^DEWHFYC]D[GSAN] | MOD_CK2_1 | 382-TTDSDPE-388 | CK2 Phosphorylation site | ...([ST])..E |
| Gene Ontology (P: Process; F: Function and C: Component terms) | (show / hide) | |
| GO:0000079 | P:regulation of cyclin-dependent protein serine/threonine kinase activity |
| GO:0001525 | P:angiogenesis |
| GO:0001933 | P:negative regulation of protein phosphorylation |
| GO:0002902 | P:regulation of B cell apoptotic process |
| GO:0004438 | F:phosphatidylinositol-3-phosphatase activity |
| GO:0004721 | F:phosphoprotein phosphatase activity |
| GO:0004722 | F:protein serine/threonine phosphatase activity |
| GO:0004725 | F:protein tyrosine phosphatase activity |
| GO:0005161 | F:platelet-derived growth factor receptor binding |
| GO:0005576 | C:extracellular region |
| GO:0005634 | C:nucleus |
| GO:0005654 | C:nucleoplasm |
| GO:0005737 | C:cytoplasm |
| GO:0005739 | C:mitochondrion |
| GO:0005829 | C:cytosol |
| GO:0005886 | C:plasma membrane |
| GO:0006470 | P:protein dephosphorylation |
| GO:0006661 | P:phosphatidylinositol biosynthetic process |
| GO:0006915 | P:apoptotic process |
| GO:0007270 | P:neuron-neuron synaptic transmission |
| GO:0007416 | P:synapse assembly |
| GO:0007417 | P:central nervous system development |
| GO:0007507 | P:heart development |
| GO:0007568 | P:aging |
| GO:0007611 | P:learning or memory |
| GO:0007613 | P:memory |
| GO:0007626 | P:locomotory behavior |
| GO:0008138 | F:protein tyrosine/serine/threonine phosphatase activity |
| GO:0008283 | P:cell proliferation |
| GO:0008284 | P:positive regulation of cell proliferation |
| GO:0008285 | P:negative regulation of cell proliferation |
| GO:0008289 | F:lipid binding |
| GO:0009749 | P:response to glucose |
| GO:0009898 | C:cytoplasmic side of plasma membrane |
| GO:0010043 | P:response to zinc ion |
| GO:0010628 | P:positive regulation of gene expression |
| GO:0010666 | P:positive regulation of cardiac muscle cell apoptotic process |
| GO:0010719 | P:negative regulation of epithelial to mesenchymal transition |
| GO:0010975 | P:regulation of neuron projection development |
| GO:0010997 | F:anaphase-promoting complex binding |
| GO:0014067 | P:negative regulation of phosphatidylinositol 3-kinase signaling |
| GO:0014823 | P:response to activity |
| GO:0016314 | F:phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase activity |
| GO:0016324 | C:apical plasma membrane |
| GO:0016477 | P:cell migration |
| GO:0016579 | P:protein deubiquitination |
| GO:0016605 | C:PML body |
| GO:0019899 | F:enzyme binding |
| GO:0021542 | P:dentate gyrus development |
| GO:0021955 | P:central nervous system neuron axonogenesis |
| GO:0030165 | F:PDZ domain binding |
| GO:0030336 | P:negative regulation of cell migration |
| GO:0030534 | P:adult behavior |
| GO:0031642 | P:negative regulation of myelination |
| GO:0031647 | P:regulation of protein stability |
| GO:0031658 | P:negative regulation of cyclin-dependent protein serine/threonine kinase activity involved in G1/S transition of mitotic cell cycle |
| GO:0032228 | P:regulation of synaptic transmission |
| GO:0032286 | GABAergic |
| GO:0032355 | P:central nervous system myelin maintenance |
| GO:0032535 | P:response to estradiol |
| GO:0032869 | P:regulation of cellular component size |
| GO:0033032 | P:cellular response to insulin stimulus |
| GO:0033198 | P:regulation of myeloid cell apoptotic process |
| GO:0033555 | P:response to ATP |
| GO:0035176 | P:multicellular organismal response to stress |
| GO:0035255 | P:social behavior |
| GO:0035749 | F:ionotropic glutamate receptor binding |
| GO:0042711 | C:myelin sheath adaxonal region |
| GO:0042802 | P:maternal behavior |
| GO:0042995 | F:identical protein binding |
| GO:0043005 | C:cell projection |
| GO:0043066 | C:neuron projection |
| GO:0043197 | P:negative regulation of apoptotic process |
| GO:0043220 | C:dendritic spine |
| GO:0043491 | C:Schmidt-Lanterman incisure |
| GO:0043542 | P:protein kinase B signaling |
| GO:0043647 | P:endothelial cell migration |
| GO:0044320 | P:inositol phosphate metabolic process |
| GO:0045211 | P:cellular response to leptin stimulus |
| GO:0045475 | C:postsynaptic membrane |
| GO:0045666 | P:locomotor rhythm |
| GO:0045792 | P:positive regulation of neuron differentiation |
| GO:0046621 | P:negative regulation of cell size |
| GO:0046685 | P:negative regulation of organ growth |
| GO:0046855 | P:response to arsenic-containing substance |
| GO:0046856 | P:inositol phosphate dephosphorylation |
| GO:0048008 | P:phosphatidylinositol dephosphorylation |
| GO:0048681 | P:platelet-derived growth factor receptor signaling pathway |
| GO:0048738 | P:negative regulation of axon regeneration |
| GO:0048853 | P:cardiac muscle tissue development |
| GO:0048854 | P:forebrain morphogenesis |
| GO:0050680 | P:brain morphogenesis |
| GO:0050765 | P:negative regulation of epithelial cell proliferation |
| GO:0050771 | P:negative regulation of phagocytosis |
| GO:0050821 | P:negative regulation of axonogenesis |
| GO:0051091 | P:protein stabilization |
| GO:0051548 | P:positive regulation of DNA-binding transcription factor activity |
| GO:0051717 | P:negative regulation of keratinocyte migration |
| GO:0051800 | F:inositol-1,3,4,5-tetrakisphosphate 3-phosphatase activity |
| GO:0051895 | F:phosphatidylinositol-3,4-bisphosphate 3-phosphatase activity |
| GO:0051898 | P:negative regulation of focal adhesion assembly |
| GO:0060024 | P:negative regulation of protein kinase B signaling |
| GO:0060044 | P:rhythmic synaptic transmission |
| GO:0060070 | P:negative regulation of cardiac muscle cell proliferation |
| GO:0060074 | P:canonical Wnt signaling pathway |
| GO:0060134 | P:synapse maturation |
| GO:0060179 | P:prepulse inhibition |
| GO:0060291 | P:male mating behavior |
| GO:0060292 | P:long-term synaptic potentiation |
| GO:0060736 | P:long term synaptic depression |
| GO:0060997 | P:prostate gland growth |
| GO:0061002 | P:dendritic spine morphogenesis |
| GO:0070373 | P:negative regulation of dendritic spine morphogenesis |
| GO:0070374 | P:negative regulation of ERK1 and ERK2 cascade |
| GO:0071257 | P:positive regulation of ERK1 and ERK2 cascade |
| GO:0071361 | P:cellular response to electrical stimulus |
| GO:0071456 | P:cellular response to ethanol |
| GO:0090071 | P:cellular response to hypoxia |
| GO:0090344 | P:negative regulation of ribosome biogenesis |
| GO:0090394 | P:negative regulation of cell aging |
| GO:0097105 | P:negative regulation of excitatory postsynaptic potential |
| GO:0097107 | P:presynaptic membrane assembly |
| GO:0099524 | P:postsynaptic density assembly |
| GO:1901017 | C:postsynaptic cytosol |
| GO:1903690 | P:negative regulation of potassium ion transmembrane transporter activity |
| GO:1903984 | P:negative regulation of wound healing |
| GO:1904668 | spreading of epidermal cells |
| GO:1904706 | P:positive regulation of TRAIL-activated apoptotic signaling pathway |
| GO:1990090 | P:positive regulation of ubiquitin protein ligase activity |
| GO:1990314 | P:negative regulation of vascular smooth muscle cell proliferation |
| GO:1990381 | P:cellular response to nerve growth factor stimulus |
| GO:1990782 | P:cellular response to insulin-like growth factor stimulus |
| GO:2000060 | F:ubiquitin-specific protease binding |
| GO:2000134 | F:protein tyrosine kinase binding |
| GO:2000272 | P:positive regulation of ubiquitin-dependent protein catabolic process |
| GO:2000463 | P:negative regulation of G1/S transition of mitotic cell cycle |
| GO:2000808 | P:negative regulation of signaling receptor activity |
Download FASTA sequences of PTEN-substrates in Humans
| Substrate Gene Name | Substrate UniProt_AC | DephosphoSite and sequence (+/-5) | Bioassay Type | PubMed ID | View in Europe PMC | Reliability of the interaction |
| SHC1 | P29353 | NA | In vitro and in vivo | 10427092, 21206972, | Europe PMC |  |
| CREB1 | P16220 | Ser-133_LSRRPsYRKIL | In vitro and in vivo | 21385900, | Europe PMC | |
| PTK2 | Q05397 | Tyr-397_SETDDyAEIID | In vitro and in vivo | 10400703, 9616126, 10427092, | Europe PMC | |
| PTK6 | Q13882 | Tyr-342_IKEDVyLSHDH | in vitro and in vivo | 29142193, | Europe PMC | |
| PDGFRB | P09619 | NA | In vitro | 14718524, | Europe PMC | |
| PTEN | P60484 | Ser-380_DHYRYsDTTDS, Thr-382_YRYSDtTDSDP, Thr-383_RYSDTtDSDPE | In vivo | 22413754, | Europe PMC | |
| Substrate ChEBI-ID | Substrate KEGG ID | DephosphoSite | Bioassay Type | PubMed ID | View in Europe PMC | Reliability of the interaction | Molecule |
| CHEBI:16618 | C05981 | D3 position | In vitro and in vivo | 9593664, 9811831, 14764604, 10555148, 18524949, | Europe PMC | |  |
| CHEBI:16152 | C11554 | D3 position | In vitro | 14764604, 10555148, 11395408, | Europe PMC | | 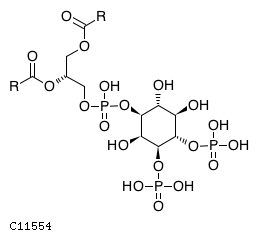 |
| CHEBI:16783 | C01272 | D3 position | In vitro | 9593664, 14764604, | Europe PMC | | 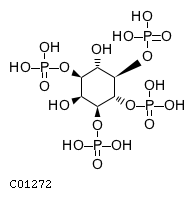 |
| CHEBI:16851 | C11556 | D3 position | In vitro | 11279206, | Europe PMC | | 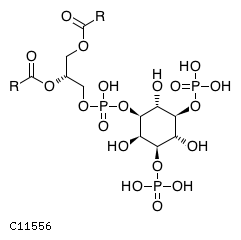 |
| CHEBI:17283 | C04549 | D3 position | In vitro | 10555148, 11395408, | Europe PMC | | 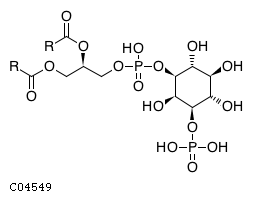 |
| Pathway ID | Pathway Name | Pathway Description (KEGG) |
| hsa00562 | Inositol phosphate metabolism | |
| hsa01100 | Metabolic pathways | |
| hsa01521 | EGFR tyrosine kinase inhibitor resistance | EGFR is a tyrosine kinase that participates in the regulation of cellular homeostasis. EGFR also serves as a stimulus for cancer growth. EGFR gene mutations and protein overexpression, both of which activate down- stream pathways, are associated with cancers, especially lung cancer. Several tyrosine kinase inhibitor (TKI) therapies against EGFR are currently administered and are initially effective in cancer patients who have EGFR mutations or aberrant activation of EGFR. However, the development of TKI resistance is common and results in the recurrence of tumors. Studies over the last decade have identified mechanisms that drive resistance to EGFR TKI treatment. Most outstanding mechanisms are: the secondary EGFR mutation (T790M), activation of alternative pathways (c-Met, HGF, AXL), aberrance of the downstream pathways (K-RAS mutations, loss of PTEN), impairment of the EGFR-TKIs-mediated apoptosis pathway (BCL2-like 11/BIM deletion polymorphism), histologic transformation, etc. |
| hsa04068 | FoxO signaling pathway | The forkhead box O (FOXO) family of transcription factors regulates the expression of genes in cellular physiological events including apoptosis, cell-cycle control, glucose metabolism, oxidative stress resistance, and longevity. A central regulatory mechanism of FOXO proteins is phosphorylation by the serine-threonine kinase Akt/protein kinase B (Akt/PKB), downstream of phosphatidylinositol 3-kinase (PI3K), in response to insulin or several growth factors. Phosphorylation at three conserved residues results in the export of FOXO proteins from the nucleus to the cytoplasm, thereby decreasing expression of FOXO target genes. In contrast, the stress-activated c-Jun N-terminal kinase (JNK) and the energy sensing AMP-activated protein kinase (AMPK), upon oxidative and nutrient stress stimuli phosphorylate and activate FoxOs. Aside from PKB, JNK and AMPK, FOXOs are regulated by multiple players through several post-translational modifications, including phosphorylation, but also acetylation, methylation and ubiquitylation. |
| hsa04070 | Phosphatidylinositol signaling system | |
| hsa04071 | Sphingolipid signaling pathway | Sphingomyelin (SM) and its metabolic products are now known to have second messenger functions in a variety of cellular signaling pathways. Particularly, the sphingolipid metabolites, ceramide (Cer) and sphingosine-1-phosphate (S1P), have emerged as a new class of potent bioactive molecules. Ceramide can be generated de novo or by hydrolysis of membrane sphingomyelin by sphingomyelinase (SMase). Ceramide is subsequently metabolized by ceramidase to generate sphingosine (Sph) which in turn produces S1P through phosphorylation by sphingosine kinases 1 and 2 (SphK1, 2). Both ceramide and S1P regulate cellular responses to stress, with generally opposing effects. S1P functions as a growth and survival factor, acting as a ligand for a family of G protein-coupled receptors, whereas ceramide activates intrinsic and extrinsic apoptotic pathways through receptor-independent mechanisms. |
| hsa04115 | p53 signaling pathway | p53 activation is induced by a number of stress signals, including DNA damage, oxidative stress and activated oncogenes. The p53 protein is employed as a transcriptional activator of p53-regulated genes. This results in three major outputs; cell cycle arrest, cellular senescence or apoptosis. Other p53-regulated gene functions communicate with adjacent cells, repair the damaged DNA or set up positive and negative feedback loops that enhance or attenuate the functions of the p53 protein and integrate these stress responses with other signal transduction pathways. |
| hsa04140 | Autophagy - animal | Autophagy (or macroautophagy) is a cellular catabolic pathway involving in protein degradation, organelle turnover, and non-selective breakdown of cytoplasmic components, which is evolutionarily conserved among eukaryotes and exquisitely regulated. This progress initiates with production of the autophagosome, a double-membrane intracellular structure of reticular origin that engulfs cytoplasmic contents and ultimately fuses with lysosomes for cargo degradation. Autophagy is regulated in response to extra- or intracellular stress and signals such as starvation, growth factor deprivation and ER stress. Constitutive level of autophagy plays an important role in cellular homeostasis and maintains quality control of essential cellular components. |
| hsa04150 | mTOR signaling pathway | The mammalian (mechanistic) target of rapamycin (mTOR) is a highly conserved serine/threonine protein kinase, which exists in two complexes termed mTOR complex 1 (mTORC1) and 2 (mTORC2). mTORC1 contains mTOR, Raptor, PRAS40, Deptor, mLST8, Tel2 and Tti1. mTORC1 is activated by the presence of growth factors, amino acids, energy status, stress and oxygen levels to regulate several biological processes, including lipid metabolism, autophagy, protein synthesis and ribosome biogenesis. On the other hand, mTORC2, which consists of mTOR, mSin1, Rictor, Protor, Deptor, mLST8, Tel2 and Tti1, responds to growth factors and controls cytoskeletal organization, metabolism and survival. |
| hsa04151 | PI3K-Akt signaling pathway | The phosphatidylinositol 3' -kinase(PI3K)-Akt signaling pathway is activated by many types of cellular stimuli or toxic insults and regulates fundamental cellular functions such as transcription, translation, proliferation, growth, and survival. The binding of growth factors to their receptor tyrosine kinase (RTK) or G protein-coupled receptors (GPCR) stimulates class Ia and Ib PI3K isoforms, respectively. PI3K catalyzes the production of phosphatidylinositol-3,4,5-triphosphate (PIP3) at the cell membrane. PIP3 in turn serves as a second messenger that helps to activate Akt. Once active, Akt can control key cellular processes by phosphorylating substrates involved in apoptosis, protein synthesis, metabolism, and cell cycle. |
| hsa04218 | Cellular senescence | Cellular senescence is a state of irreversible cellular arrest and can be triggered by a number of factors, such as telomere shortening, oncogene activation, irradiation, DNA damage and oxidative stress. It is characterized by enlarged flattened morphology, senescence-associated beta-galactosidase (SA-b-gal) activity, secretion of inflammatory cytokines, growth factors and matrix metalloproteinases, as part of the senescence-associated secretory phenotype (SASP). Cellular senescence is functionally associated with many biological processes including aging, tumor suppression, placental biology, embryonic development, and wound healing. |
| hsa04510 | Focal adhesion | Cell-matrix adhesions play essential roles in important biological processes including cell motility, cell proliferation, cell differentiation, regulation of gene expression and cell survival. At the cell-extracellular matrix contact points, specialized structures are formed and termed focal adhesions, where bundles of actin filaments are anchored to transmembrane receptors of the integrin family through a multi-molecular complex of junctional plaque proteins. Some of the constituents of focal adhesions participate in the structural link between membrane receptors and the actin cytoskeleton, while others are signalling molecules, including different protein kinases and phosphatases, their substrates, and various adapter proteins. Integrin signaling is dependent upon the non-receptor tyrosine kinase activities of the FAK and src proteins as well as the adaptor protein functions of FAK, src and Shc to initiate downstream signaling events. These signalling events culminate in reorganization of the actin cytoskeleton; a prerequisite for changes in cell shape and motility, and gene expression. Similar morphological alterations and modulation of gene expression are initiated by the binding of growth factors to their respective receptors, emphasizing the considerable crosstalk between adhesion- and growth factor-mediated signalling. |
| hsa04931 | Insulin resistance | Insulin resistance is a condition where cells become resistant to the effects of insulin. It is often found in people with health disorders, including obesity, type 2 diabetes mellitus, non-alcoholic fatty liver disease, and cardiovascular diseases. In this diagram multiple mechanisms underlying insulin resistance are shown: (a) increased phosphorylation of IRS (insulin receptor substrate) protein through serine/threonine kinases, such as JNK1 and IKKB, and protein kinase C, (b) increased IRS-1 proteasome degradation via mTOR signaling pathway, (c) decreased activation of signaling molecules including PI3K and AKT, (d) increase in activity of phosphatases including PTPs, PTEN, and PP2A. Regulatory actions such as oxidative stress, mitochondrial dysfunction, accumulation of intracellular lipid derivatives (diacylglycrol and ceramides), and inflammation (via IL-6 and TNFA) contribute to these mechanisms. Consequently, insulin resistance causes reduced GLUT4 translocation, resulting in glucose takeup and glycogen synthesis in skeletal muscle as well as increased hepatic gluconeogenesis and decreased glycogen synthesis in liver. At the bottom of the diagram, interplay between O-GlcNAcylation and serine/threonine phosphorylation is shown. Studies suggested that elevated O-GlcNAc level was correlated to high glucose-induced insulin resistance. Donor UDP-GlcNAc is induced through hexosamine biosynthesis pathway and added to proteins by O-GlcNAc transferase. Elevation of O-GlcNAc modification alters phosphorylation and function of key insulin signaling proteins including IRS-1, PI3K, PDK1, Akt and other transcription factor and cofactors, resulting in the attenuation of insulin signaling cascade. |
| hsa05161 | Hepatitis B | Hepatitis B virus (HBV) is an enveloped virus and contains a partially double-stranded relaxed circular DNA (RC-DNA) genome. After entry into hepatocytes, HBV RC-DNA is transported to the nucleus and converted into a covalently closed circular molecule cccDNA. The cccDNA is the template for transcription of all viral RNAs including the pregenomic RNA (pgRNA), encoding for 7 viral proteins: large, middle, and small envelope proteins (LHBs, MHBs, and SHBs) that form the surface antigen (HBsAg), the core antigen (HBcAg), the e antigen (HBeAg), the HBV polymerase, and the regulatory protein X (HBx). The pgRNA interacts with the viral polymerase protein to initiate the encapsidation into the core particles. Through endoplasmic reticulum, the core particles finish assembling with the envelope proteins and are released. HBV infection leads to a wide spectrum of liver diseases raging from chronic hepatitis, cirrhosis to hepatocellular carcinoma. The mechanism of liver injury is still not clear. However, HBV proteins target host proteins, involved in a variety of functions, thus regulating transcription, cellular signaling cascades, proliferation, differentiation, and apoptosis. |
| hsa05165 | Human papillomavirus infection | Human papillomavirus (HPV) is a non-enveloped, double-stranded DNA virus. HPV infect mucoal and cutaneous epithelium resulting in several types of pathologies, most notably, cervical cancer. All types of HPV share a common genomic structure and encode eight proteins: E1, E2, E4, E5, E6, and E7 (early) and L1 and L2 (late). It has been demonstrated that E1 and E2 are involved in viral transcription and replication. The functions of the E4 protein is not yet fully understood. E5, E6, and E7 act as oncoproteins. E5 inhibits the V-ATPase, prolonging EGFR signaling and thereby promoting cell proliferation. The expression of E6 and E7 not only inhibits the tumor suppressors p53 and Rb, but also alters additional signalling pathways. Among these pathways, PI3K/Akt signalling cascade plays a very important role in HPV-induced carcinogenesis. The L1 and L2 proteins form icosahedral capsids for progeny virion generation. |
| hsa05200 | Pathways in cancer | |
| hsa05206 | MicroRNAs in cancer | MicroRNA (miRNA) is a cluster of small non-encoding RNA molecules of 21 - 23 nucleotides in length, which controls gene expression post-transcriptionally either via the degradation of target mRNAs or the inhibition of protein translation. Using high-throughput profiling, dysregulation of miRNAs has been widely observed in different stages of cancer. The upregulation (overexpression) of specific miRNAs could lead to the repression of tumor suppressor gene expression, and conversely the downregulation of specific miRNAs could result in an increase of oncogene expression; both these situations induce subsequent malignant effects on cell proliferation, differentiation, and apoptosis that lead to tumor growth and progress. The miRNA signatures of cancer observed in various studies differ significantly. These inconsistencies occur due to the differences in the study populations and methodologies used. This pathway map shows the summarized results from various studies in 9 cancers, each of which is presented in a review article. |
| hsa05213 | Endometrial cancer | Endometrial cancer (EC) is the most common gynaecological malignancy and the fourth most common malignancy in women in the developed world after breast, colorectal and lung cancer. Two types of endometrial carcinoma are distinguished with respect to biology and clinical course. Type-I carcinoma is related to hyperestrogenism by association with endometrial hyperplasia, frequent expression of estrogen and progesterone receptors and younger age, whereas type-II carcinoma is unrelated to estrogen, associated with atrophic endometrium, frequent lack of estrogen and progesterone receptors and older age. The morphologic differences in these cancers are mirrored in their molecular genetic profile with type I showing defects in DNA-mismatch repair and mutations in PTEN, K-ras, and beta-catenin, and type II showing aneuploidy, p53 mutations, and her2/neu amplification. |
| hsa05214 | Glioma | Gliomas are the most common of the primary brain tumors and account for more than 40% of all central nervous system neoplasms. Gliomas include tumours that are composed predominantly of astrocytes (astrocytomas), oligodendrocytes (oligodendrogliomas), mixtures of various glial cells (for example,oligoastrocytomas) and ependymal cells (ependymomas). The most malignant form of infiltrating astrocytoma - glioblastoma multiforme (GBM) - is one of the most aggressive human cancers. GBM may develop de novo (primary glioblastoma) or by progression from low-grade or anaplastic astrocytoma (secondary glioblastoma). Primary glioblastomas develop in older patients and typically show genetic alterations (EGFR amplification, p16/INK4a deletion, and PTEN mutations) at frequencies of 24-34%. Secondary glioblastomas develop in younger patients and frequently show overexpression of PDGF and CDK4 as well as p53 mutations (65%) and loss of Rb playing major roles in such transformations. Loss of PTEN has been implicated in both pathways, although it is much more common in the pathogenesis of primary GBM. |
| hsa05215 | Prostate cancer | Prostate cancer constitutes a major health problem in Western countries. It is the most frequently diagnosed cancer among men and the second leading cause of male cancer deaths. The identification of key molecular alterations in prostate-cancer cells implicates carcinogen defenses (GSTP1), growth-factor-signaling pathways (NKX3.1, PTEN, and p27), and androgens (AR) as critical determinants of the phenotype of prostate-cancer cells. Glutathione S-transferases (GSTP1) are detoxifying enzymes. Cells of prostatic intraepithelial neoplasia, devoid of GSTP1, undergo genomic damage mediated by carcinogens. NKX3.1, PTEN, and p27 regulate the growth and survival of prostate cells in the normal prostate. Inadequate levels of PTEN and NKX3.1 lead to a reduction in p27 levels and to increased proliferation and decreased apoptosis. Androgen receptor (AR) is a transcription factor that is normally activated by its androgen ligand. During androgen withdrawal therapy, the AR signal transduction pathway also could be activated by amplification of the AR gene, by AR gene mutations, or by altered activity of AR coactivators. Through these mechanisms, tumor cells lead to the emergence of androgen-independent prostate cancer. |
| hsa05218 | Melanoma | Melanoma is a form of skin cancer that has a poor prognosis and which is on the rise in Western populations. Melanoma arises from the malignant transformation of pigment-producing cells, melanocytes. The only known environmental risk factor is exposure to ultraviolet (UV) light and in people with fair skin the risk is greatly increased. Melanoma pathogenesis is also driven by genetic factors. Oncogenic NRAS mutations activate both effector pathways Raf-MEK-ERK and PI3K-Akt. The Raf-MEK-ERK pathway may also be activated via mutations in the BRAF gene. The PI3K-Akt pathway may be activated through loss or mutation of the inhibitory tumor suppressor gene PTEN. These mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Melanoma development has been shown to be strongly associated with inactivation of the p16INK4a/cyclin dependent kinases 4 and 6/retinoblastoma protein (p16INK4a/CDK4,6/pRb) and p14ARF/human double minute 2/p53 (p14ARF/HMD2/p53) tumor suppressor pathways. MITF and TP53 are implicated in further melanoma progression. |
| hsa05222 | Small cell lung cancer | Lung cancer is a leading cause of cancer death among men and women in industrialized countries. Small cell lung carcinoma (SCLC) is a highly aggressive neoplasm, which accounts for approximately 25% of all lung cancer cases. Molecular mechanisms altered in SCLC include induced expression of oncogene, MYC, and loss of tumorsuppressor genes, such as p53, PTEN, RB, and FHIT. The overexpression of MYC proteins in SCLC is largely a result of gene amplification. Such overexpression leads to more rapid proliferation and loss of terminal differentiation. Mutation or deletion of p53 or PTEN can lead to more rapid proliferation and reduced apoptosis. The retinoblastoma gene RB1 encodes a nuclear phosphoprotein that helps to regulate cell-cycle progression. The fragile histidine triad gene FHIT encodes the enzyme diadenosine triphosphate hydrolase, which is thought to have an indirect role in proapoptosis and cell-cycle control. |
| hsa05224 | Breast cancer | Breast cancer is the leading cause of cancer death among women worldwide. The vast majority of breast cancers are carcinomas that originate from cells lining the milk-forming ducts of the mammary gland. The molecular subtypes of breast cancer, which are based on the presence or absence of hormone receptors (estrogen and progesterone subtypes) and human epidermal growth factor receptor-2 (HER2), include: hormone receptor positive and HER2 negative (luminal A subtype), hormone receptor positive and HER2 positive (luminal B subtype), hormone receptor negative and HER2 positive (HER2 positive), and hormone receptor negative and HER2 negative (basal-like or triple-negative breast cancers (TNBCs)). Hormone receptor positive breast cancers are largely driven by the estrogen/ER pathway. In HER2 positive breast tumours, HER2 activates the PI3K/AKT and the RAS/RAF/MAPK pathways, and stimulate cell growth, survival and differentiation. In patients suffering from TNBC, the deregulation of various signalling pathways (Notch and Wnt/beta-catenin), EGFR protein have been confirmed. In the case of breast cancer only 8% of all cancers are hereditary, a phenomenon linked to genetic changes in BRCA1 or BRCA2. Somatic mutations in only three genes (TP53, PIK3CA and GATA3) occurred at >10% incidence across all breast cancers. |
| hsa05225 | Hepatocellular carcinoma | Hepatocellular carcinoma (HCC) is a major type of primary liver cancer and one of the rare human neoplasms etiologically linked to viral factors. It has been shown that, after HBV/HCV infection and alcohol or aflatoxin B1 exposure, genetic and epigenetic changes occur. The recurrent mutated genes were found to be highly enriched in multiple key driver signaling processes, including telomere maintenance, TP53, cell cycle regulation, the Wnt/beta-catenin pathway (CTNNB1 and AXIN1), the phosphatidylinositol-3 kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway. Recent studies using whole-exome sequencing have revealed recurrent mutations in new driver genes involved in the chromatin remodelling (ARID1A and ARID2) and the oxidative stress (NFE2L2) pathways. |
| hsa05230 | Central carbon metabolism in cancer | Malignant transformation of cells requires specific adaptations of cellular metabolism to support growth and survival. In the early twentieth century, Otto Warburg established that there are fundamental differences in the central metabolic pathways operating in malignant tissue. He showed that cancer cells consume a large amount of glucose, maintain high rate of glycolysis and convert a majority of glucose into lactic acid even under normal oxygen concentrations (Warburg's Effects). More recently, it has been recognized that the 'Warburg effect' encompasses a similarly increased utilization of glutamine. From the intermediate molecules provided by enhanced glycolysis and glutaminolysis, cancer cells synthesize most of the macromolecules required for the duplication of their biomass and genome. These cancer-specific alterations represent a major consequence of genetic mutations and the ensuing changes of signalling pathways in cancer cells. Three transcription factors, c-MYC, HIF-1 and p53, are key regulators and coordinate regulation of cancer metabolism in different ways, and many other oncogenes and tumor suppressor genes cluster along the signaling pathways that regulate c-MYC, HIF-1 and p53. |
| Pathway ID | Pathway Name | Pathway Description (KEGG) |
| R-HSA-1660499 | Synthesis of PIPs at the plasma membrane. | At the plasma membrane, subsequent phosphorylation of phosphatidylinositol 4-phosphate (PI4P) produces phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) and phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3) while the actions of various other kinases and phosphatases produces phosphatidylinositol 3-phosphate (PI3P), phosphatidylinositol 5-phosphate (PI5P), phosphatidylinositol 3,4-bisphosphate (PI(3,4)P2), and phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) (Zhang et al. 1997, Gurung et al. 2003, Guo et al. 1999, Vanhaesebroeck et al. 1997, Tolias et al. 1998, Schaletzky et al. 2003, Kim et al. 2002, Clarke et al. 2010). Many of the phosphatidylinositol phosphatases that act at the plasma membrane belong to the myotubularin family. Enzymatically inactive myotubularin family members can heterodimerize with catalytically active mytotubularins to regulate their stability, activity and/or substrate specificity (Berger et al. 2006, Zou et al. 2012) |
| R-HSA-1855204 | Synthesis of IP3 and IP4 in the cytosol. | An array of inositol trisphosphate (IP3) and tetrakisphosphate (IP4) molecules are synthesised by the action of various kinases and phosphatases in the cytosol (Irvine & Schell 2001, Bunney & Katan 2010) |
| R-HSA-199418 | Negative regulation of the PI3K/AKT network. | The PI3K/AKT network is negatively regulated by phosphatases that dephosphorylate PIP3, thus hampering AKT activation |
| R-HSA-202424 | Downstream TCR signaling. | Changes in gene expression are required for the T cell to gain full proliferative competence and to produce effector cytokines. Three transcription factors in particular have been found to play a key role in TCR-stimulated changes in gene expression, namely NF-kB, NFAT and AP-1. A key step in NF-kB activation is the stimulation and translocation of PKC theta. The critical element that effects PKC theta activation is PI3K. This enzyme complex translocates to the plasma membrane by interacting with phospho-tyrosines on CD28 via its two SH2 domains located in p85 subunit. The p110 subunit of PI3K phosphorylates the inositol ring of PIP2 to generate PIP3 (steps 17-18). PIP3 may also be dephosphorylated by the phosphatase SHIP to generate PI-3,4-P2. PIP3 and PI-3,4-P2 acts as binding sites to the PH domain of PKB/Akt and PDK1 (steps 19, 21 and 22). PKB is activated in response to PI3K stimulation by PDK1 (step 23). PDK1 has an essential role in regulating the activation of PKC theta and recruitment of CBM complex to the immune synapse. PKC theta is a member of novel class (DAG dependent, Ca++ independent) of PKC and the only member known to translocate to this synapse. Prior to TCR stimulation PKC theta exists in an inactive closed conformation. Upon release of DAG, it binds to PKC theta via the C1 domain and undergoes phosphorylation on tyrosine 90 by Lck to attain an open conformation. PKC theta is further phosphorylated by PDK1 on threonine 538. This step is critical for PKC activity (steps 24-26). CARMA1 translocates to the plasma membrane following the interaction of its SH3 domain with the 'PxxP' motif on PDK1. CARMA1 is phosphorylated by PKC-theta on residue S552, leading to the oligomerization of CARMA1. This complex acts as a scaffold, recruiting Bcl10 to the synapse by interacting with their CARD domains. Bcl10 undergoes phosphorylation mediated by the enzyme RIP2. Activated Bcl10 then mediates the ubiquitination of NEMO by recruiting MALT1 and TRAF6. MALT1 binds to Bcl10 with its Ig-like domains and undergoes oligomerization. TRAF6 binds to the oligomerized MALT1 and also undergoes oligomerization. Oligomerized TRAF6 acts as a ubiquitin-protein ligase, catalyzing auto-K63-linked polyubiquitination (steps 27-33). This K-63 ubiquitinated TRAF6 activates TAK1 kinase bound to TAB2 and also ubiquitinates NEMO/IKK-gamma in the IKK complex. TAK1 undergoes autophosphorylation on residues T184 and T187 and gets activated. Activated TAK1 kinase phosphorylates IKK-beta on residues S177 and S181 in the activation loop and activates the IKK kinase activity. IKK-beta phosphorylates the IkB-alpha bound to the NF-kB heterodimer, on residues S19 and S23 and directs IkB-beta to 26S proteasome degradation (step 34-38 & 40). The NF-kB heterodimer with a free NTS sequence finally migrates to the nucleus to regulate gene transcription (step 39) |
| R-HSA-5628897 | TP53 Regulates Metabolic Genes. | While the p53 tumor suppressor protein (TP53) is known to inhibit cell growth by inducing apoptosis, senescence and cell cycle arrest, recent studies have found that p53 is also able to influence cell metabolism to prevent tumor development. TP53 regulates transcription of many genes involved in the metabolism of carbohydrates, nucleotides and amino acids, protein synthesis and aerobic respiration. TP53 stimulates transcription of TIGAR, a D-fructose 2,6-bisphosphatase. TIGAR activity decreases glycolytic rate and lowers ROS (reactive oxygen species) levels in cells (Bensaad et al. 2006). TP53 may also negatively regulate the rate of glycolysis by inhibiting the expression of glucose transporters GLUT1, GLUT3 and GLUT4 (Kondoh et al. 2005, Schwartzenberg-Bar-Yoseph et al. 2004, Kawauchi et al. 2008). TP53 negatively regulates several key points in PI3K/AKT signaling and downstream mTOR signaling, decreasing the rate of protein synthesis and, hence, cellular growth. TP53 directly stimulates transcription of the tumor suppressor PTEN, which acts to inhibit PI3K-mediated activation of AKT (Stambolic et al. 2001). TP53 stimulates transcription of sestrin genes, SESN1, SESN2, and SESN3 (Velasco-Miguel et al. 1999, Budanov et al. 2002, Brynczka et al. 2007). One of sestrin functions may be to reduce and reactivate overoxidized peroxiredoxin PRDX1, thereby reducing ROS levels (Budanov et al. 2004, Papadia et al. 2008, Essler et al. 2009). Another function of sestrins is to bind the activated AMPK complex and protect it from AKT-mediated inactivation. By enhancing AMPK activity, sestrins negatively regulate mTOR signaling (Budanov and Karin 2008, Cam et al. 2014). The expression of DDIT4 (REDD1), another negative regulator of mTOR signaling, is directly stimulated by TP63 and TP53. DDIT4 prevents AKT-mediated inactivation of TSC1:TSC2 complex, thus inhibiting mTOR cascade (Cam et al. 2014, Ellisen et al. 2002, DeYoung et al. 2008). TP53 may also be involved, directly or indirectly, in regulation of expression of other participants of PI3K/AKT/mTOR signaling, such as PIK3CA (Singh et al. 2002), TSC2 and AMPKB (Feng et al. 2007). TP53 regulates mitochondrial metabolism through several routes. TP53 stimulates transcription of SCO2 gene, which encodes a mitochondrial cytochrome c oxidase assembly protein (Matoba et al. 2006). TP53 stimulates transcription of RRM2B gene, which encodes a subunit of the ribonucleotide reductase complex, responsible for the conversion of ribonucleotides to deoxyribonucleotides and essential for the maintenance of mitochondrial DNA content in the cell (Tanaka et al. 2000, Bourdon et al. 2007, Kulawiec et al. 2009). TP53 also transactivates mitochondrial transcription factor A (TFAM), a nuclear-encoded gene important for mitochondrial DNA (mtDNA) transcription and maintenance (Park et al. 2009). Finally, TP53 stimulates transcription of the mitochondrial glutaminase GLS2, leading to increased mitochondrial respiration rate and reduced ROS levels (Hu et al. 2010). The great majority of tumor cells generate energy through aerobic glycolysis, rather than the much more efficient aerobic mitochondrial respiration, and this metabolic change is known as the Warburg effect (Warburg 1956). Since the majority of tumor cells have impaired TP53 function, and TP53 regulates a number of genes involved in glycolysis and mitochondrial respiration, it is likely that TP53 inactivation plays an important role in the metabolic derangement of cancer cells such as the Warburg effect and the concomitant increased tumorigenicity (reviewed by Feng and Levine 2010). On the other hand, some mutations of TP53 in Li-Fraumeni syndrome may result in the retention of its wild-type metabolic activities while losing cell cycle and apoptosis functions (Wang et al. 2013). Consistent with such human data, some mutations of p53, unlike p53 null state, retain the ability to regulate energy metabolism while being inactive in regulating its classic gene targets involved in cell cycle, apoptosis and senescence. Retention of metabolic and antioxidant functions of p53 protects p53 mutant mice from early onset tumorigenesis (Li et al. 2012) |
| R-HSA-5674404 | PTEN Loss of Function in Cancer. | Loss-of-function mutations affecting the phosphatase domain of PTEN are frequently found in sporadic cancers (Kong et al. 1997, Lee et al. 1999, Han et al. 2000), as well as in PTEN hamartoma tumor syndromes (PHTS) (Marsh et al. 1998). PTEN can also be inactivated by gene deletion or epigenetic silencing, or indirectly by overexpression of microRNAs that target PTEN mRNA (Huse et al. 2009). Cells with deficient PTEN function have increased levels of PIP3, and therefore increased AKT activity. For a recent review, please refer to Hollander et al. 2011 |
| R-HSA-5689880 | Ub-specific processing proteases. | Ub-specific processing proteases (USPs) are the largest of the DUB families with more than 50 members in humans. The USP catalytic domain varies considerably in size and consists of six conserved motifs with N- or C-terminal extensions and insertions occurring between the conserved motifs (Ye et al. 2009). Two highly conserved regions comprise the catalytic triad, the Cys-box (Cys) and His-box (His and Asp/Asn) (Nijman et al. 2005, Ye et al. 2009, Reyes-Turcu & Wilkinson 2009). They recognize their substrates by interactions of the variable regions with the substrate protein directly, or via scaffolds or adapters in multiprotein complexes |
| R-HSA-5689896 | Ovarian tumor domain proteases. | Humans have 16 Overian tumour domain (OTU) family DUBs that can be evolutionally divided into three classes, the OTUs, the Otubains (OTUBs), and the A20-like OTUs (Komander et al. 2009). OTU family DUBs can be highly selective in the type of ubiquitin crosslinks they cleave. OTUB1 is specific for K48-linked chains, whereas OTUB2 can cleave K11, K63 and K48-linked poly-Ub (Wang et al. 2009, Edelmann et al. 2009, Mevissen et al. 2013). A20 prefers K48-linked chains, Cezanne is specific for K11-linked chains, and TRABID acts on both K29, K33 and K63-linked poly-Ub (Licchesi et al. 2011, Komander & Barford 2008, Bremm et al. 2010, Mevissen et al. 2013). The active site of the OTU domain contains an unusual loop not seen in other thiol-DUBs and can lack an obvious catalytic Asp/Asn (Komander & Barford 2009, Messick et al. 2008, Lin et al. 2008). A20 and OTUB1 have an unusual mode of activity, binding directly to E2 enzymes (Nakada et al. 2010, Wertz et al. 2004) |
| R-HSA-8943723 | Regulation of PTEN mRNA translation. | MicroRNAs miR-17, miR-19a, miR-19b, miR-20a, miR-20b, miR-21, miR-22, miR-25, miR 26A1, miR 26A2, miR-93, miR-106a, miR-106b, miR 205, and miR 214 and bind PTEN mRNA and inhibit its translation into protein. These microRNAs are altered in cancer and can account for changes in PTEN levels. There is evidence that PTEN mRNA translation is also inhibited by other microRNAs, such as miR-302 and miR-26B, and these microRNAs will be annotated when additional experimental details become available (Meng et al. 2007, Xiao et al. 2008, Yang et al. 2008, Huse et al. 2009, Kim et al. 2010, Poliseno, Salmena, Riccardi et al. 2010, Zhang et al. 2010, Tay et al. 2011, Qu et al. 2012, Cai et al. 2013). In addition, coding and non coding RNAs can prevent microRNAs from binding to PTEN mRNA. These RNAs are termed competing endogenous RNAs or ceRNAs. Transcripts of the pseudogene PTENP1 and mRNAs transcribed from SERINC1, VAPA and CNOT6L genes exhibit this activity (Poliseno, Salmena, Zhang et al. 2010, Tay et al. 2011, Tay et al. 2014) |
| R-HSA-8948747 | Regulation of PTEN localization. | When monoubiquitinated by E3 ubiquitin ligases XIAP and NEDD4, PTEN translocates from the cytosol to the nucleus (Trotman et al. 2007, Van Themsche et al. 2009). USP7 (HAUSP)-mediated deubiquitination of monoubiquitinated nuclear PTEN promotes relocalization of PTEN to the cytosol (Song et al. 2008) |
| R-HSA-8948751 | Regulation of PTEN stability and activity. | PTEN protein stability is regulated by ubiquitin ligases, such as NEDD4, WWP2, STUB1 (CHIP), XIAP, MKRN1 and RNF146, which polyubiquitinate PTEN in response to different stimuli and thus target it for proteasome-mediated degradation (Wang et al. 2007, Van Themsche et al. 2009, Maddika et al. 2011, Ahmed et al. 2012, Lee et al. 2015, Li et al. 2015). Several ubiquitin proteases, such as USP13 and OTUD3, can remove polyubiquitin chains from PTEN and rescue it from degradation (Zhang et al. 2013, Yuan et al. 2015). TRIM27 (RFP) is an E3 ubiquitin ligase that polyubiquitinates PTEN on multiple lysines in the C2 domain of PTEN using K27 linkage between ubiquitin molecules. TRIM27 mediated ubiquitination inhibits PTEN lipid phosphatase activity, but does not affect PTEN protein localization or stability (Lee et al. 2013).PTEN phosphorylation by the tyrosine kinase FRK (RAK) inhibits NEDD4 mediated polyubiquitination and subsequent degradation of PTEN, thus increasing PTEN half life. FRK mediated phosphorylation also increases PTEN enzymatic activity (Yim et al. 2009). Casein kinase 2 (CK2) mediated phosphorylation of the C-terminus of PTEN on multiple serine and threonine residues increases PTEN protein stability (Torres and Pulido 2001) but results in ~30% reduction in PTEN lipid phosphatase activity (Miller et al. 2002).PREX2, a RAC1 guanine nucleotide exchange factor (GEF) can binds to PTEN and inhibit its catalytic activity (Fine et al. 2009) |
| Interacting partner | Detection method | Interaction type | PubMed IDs | Interaction Score | Source |
| ACACA | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| ACACB | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| ADA | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| ADGRF3 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| AKAP12 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| AKT1 | Affinity Capture-Western, genetic interference | genetic interaction, physical | 20596030, 20605778, (Europe PMC) | 0.17 | BioGRID, IntAct, MINT |
| AMHR2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| AMOT | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| ANAPC4 | Affinity Capture-MS, Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation | association, physical | 21241890, (Europe PMC) | 0.46 | BioGRID, IntAct |
| ANAPC5 | Affinity Capture-MS, Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation | association, physical | 21241890, (Europe PMC) | 0.46 | BioGRID, IntAct |
| ANAPC7 | Affinity Capture-MS, Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation | association, physical | 21241890, (Europe PMC) | 0.46 | BioGRID, IntAct |
| ANG | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| ANXA2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| AR | Affinity Capture-Western, Reconstituted Complex | physical | 15205473, (Europe PMC) | NA | BioGRID |
| ARF4 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| ARHGAP26 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| ATP5F1A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| ATP5F1C | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| BAP1 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| BCAR1 | Reconstituted Complex | physical | 9927060, (Europe PMC) | NA | BioGRID |
| BCKDHA | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| BEX1 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| BLOC1S1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| BMI1 | Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, cross-linking study, fluorescence microscopy | colocalization, physical, physical association | 19903340, (Europe PMC) | 0.61 | BioGRID, IntAct, MINT |
| BRAF | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| BRD4 | Negative Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| CALM1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| CARS2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| CASP8 | Affinity Capture-MS, Affinity Capture-Western | physical | 15717329, (Europe PMC) | NA | BioGRID |
| CAV1 | Affinity Capture-Western, coimmunoprecipitation, cosedimentation through density gradient | colocalization, physical, physical association | 12176037, (Europe PMC) | 0.50 | BioGRID, IntAct, MINT |
| CBL | Affinity Capture-Western | physical | 23118026, (Europe PMC) | NA | BioGRID |
| CCNE2 | Affinity Capture-MS, Affinity Capture-Western | physical | 15717329, (Europe PMC) | NA | BioGRID |
| CDC27 | Affinity Capture-MS, Affinity Capture-Western, Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, fluorescence microscopy | association, colocalization, physical, physical association | 21241890, (Europe PMC) | 0.62 | BioGRID, IntAct |
| CDC42EP5 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| CDH1 | Affinity Capture-Western | physical | 24811168, (Europe PMC) | NA | BioGRID |
| CDK4 | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| CDKN2A | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| CENPC | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| CFB | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| CFL1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| CHEK1 | Negative Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| CHGB | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| COPS6 | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| CRKL | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| CSNK2A1 | Affinity Capture-MS, Biochemical Activity, Reconstituted Complex | physical | 12297295, 15717329, 25047839, (Europe PMC) | NA | BioGRID |
| CSNK2A2 | Biochemical Activity, Reconstituted Complex | physical | 12297295, (Europe PMC) | NA | BioGRID |
| CTNNB1 | Affinity Capture-Western, anti bait coimmunoprecipitation | physical, physical association | 20123964, 20372837, (Europe PMC) | 0.40 | BioGRID, IntAct |
| CXCL1 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| CYB5R3 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| DBF4B | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| DBN1 | Reconstituted Complex, anti tag coimmunoprecipitation, fluorescent resonance energy transfer | physical, physical association | 23940795, 26561776, (Europe PMC) | 0.52 | BioGRID, IntAct |
| DBT | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| DCAF11 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| DDB1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| DLC1 | Affinity Capture-Western, Reconstituted Complex | physical | 19482022, (Europe PMC) | NA | BioGRID |
| DLG1 | Reconstituted Complex | physical | 11439092, (Europe PMC) | NA | BioGRID |
| DNAJA1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| DSP | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| EEF1A2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| EEF2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| EGFR | Affinity Capture-Western | physical | 26531778, (Europe PMC) | NA | BioGRID |
| EGR1 | Affinity Capture-Western | physical | 21354147, (Europe PMC) | NA | BioGRID |
| EIF2AK2 | Affinity Capture-Western | physical | 20029030, (Europe PMC) | NA | BioGRID |
| ELAVL1 | Affinity Capture-RNA | physical | 19322201, (Europe PMC) | NA | BioGRID |
| ELOA3 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| EME2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| ENO2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| EPHB1 | Affinity Capture-Western | physical | 23118026, (Europe PMC) | NA | BioGRID |
| ERBB3 | Two-hybrid | physical | 28065597, (Europe PMC) | NA | BioGRID |
| ESR1 | Reconstituted Complex | physical | 15205473, (Europe PMC) | NA | BioGRID |
| ETS1 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| FASN | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| FBN2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| FBXL6 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| FBXW7 | Affinity Capture-Western | physical | 23514585, (Europe PMC) | NA | BioGRID |
| FLNB | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| FRK | Affinity Capture-MS, Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, protein kinase assay, pull down | phosphorylation reaction, physical, physical association | 19345329, (Europe PMC) | 0.64 | BioGRID, IntAct |
| FZR1 | Affinity Capture-Western, Reconstituted Complex | physical | 21241890, 24811168, (Europe PMC) | NA | BioGRID |
| G3BP2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| GFRA2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| GNAI1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| GNAI2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| GNB4 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| GPC1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| GPC4 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| GPER1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| GSTM2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| HAGHL | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| HBA1 | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| HEATR6 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| HES5 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| HES6 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| HIST1H2AB | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| HNRNPL | Affinity Capture-RNA | physical | 28611215, (Europe PMC) | NA | BioGRID |
| HNRNPU | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| HSD17B1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| HSP90AA1 | Affinity Capture-MS, Positive Genetic | genetic, physical | 21532586, 28319113, (Europe PMC) | NA | BioGRID |
| HSP90AB1 | Affinity Capture-MS, Reconstituted Complex | physical | 21532586, 26561776, (Europe PMC) | NA | BioGRID |
| HSPA1A | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| HSPA1L | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| HSPA2 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| HSPA6 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| HSPA8 | Affinity Capture-MS, Reconstituted Complex | physical | 21532586, 26561776, (Europe PMC) | NA | BioGRID |
| HSPD1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| IFIH1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| IL24 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| INHBE | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| INS | Biochemical Activity | physical | 11439092, (Europe PMC) | NA | BioGRID |
| IRS4 | Affinity Capture-MS, Affinity Capture-Western | physical | 15717329, (Europe PMC) | NA | BioGRID |
| ITPR3 | Affinity Capture-Western | physical | 28614300, (Europe PMC) | NA | BioGRID |
| ITPRID2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| JUP | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| KAT2B | Affinity Capture-Western | physical | 16829519, (Europe PMC) | NA | BioGRID |
| KBTBD4 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| KDM6A | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| KRT14 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| LATS1 | Biochemical Activity | physical | 22641346, (Europe PMC) | NA | BioGRID |
| LDHB | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| LGALS1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| LIMA1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| LLGL2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| MAGI3 | Affinity Capture-Western, Reconstituted Complex, Two-hybrid, anti tag coimmunoprecipitation, pull down, two hybrid | direct interaction, physical, physical association | 10748157, (Europe PMC) | 0.63 | BioGRID, IntAct, MINT |
| MAP2K6 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| MCCC1 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| MCCC2 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| MCRS1 | Affinity Capture-Western, Far Western, Reconstituted Complex | physical | 15659546, (Europe PMC) | NA | BioGRID |
| MED12 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| MET | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| METTL26 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MPRIP | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MTHFD2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MTOR | Negative Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| MVP | Affinity Capture-Western, Reconstituted Complex, Two-hybrid | physical | 12177006, (Europe PMC) | NA | BioGRID |
| MXD1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| MYH10 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MYL12A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MYO1B | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MYO1C | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MYO1D | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MYOF | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| NCF1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| NCOA3 | Affinity Capture-Western, anti bait coimmunoprecipitation | physical, physical association | 23514585, (Europe PMC) | 0.40 | BioGRID, IntAct |
| NDFIP1 | Affinity Capture-Western, PCA | physical | 20534535, 22213801, 24798731, (Europe PMC) | NA | BioGRID |
| NDFIP2 | Affinity Capture-Western | physical | 20534535, (Europe PMC) | NA | BioGRID |
| NEDD4 | Affinity Capture-Western, Biochemical Activity, Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | physical, physical association | 18307411, 18498243, 19345329, 21986318, 24063548, 24141722, 25047839, 25547115, 27295432, (Europe PMC) | 0.59 | BioGRID, IntAct |
| NF1 | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| NGB | Reconstituted Complex | physical | 23904011, (Europe PMC) | NA | BioGRID |
| NME1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| NOTCH1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| OPRL1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| OSBP2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| OSGIN1 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| OTUD3 | Affinity Capture-Western, Biochemical Activity, Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | physical, physical association | 26280536, (Europe PMC) | 0.59 | BioGRID, IntAct |
| P3H4 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PARK7 | Affinity Capture-Western, Reconstituted Complex | physical | 19885556, 21426932, (Europe PMC) | NA | BioGRID |
| PBK | Affinity Capture-Western, Biochemical Activity | physical | 24012691, (Europe PMC) | NA | BioGRID |
| PBX3 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| PC | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| PCCA | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| PCCB | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| PDGFRA | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PDGFRB | Co-localization, filter binding, proximity ligation assay, pull down | association, direct interaction, physical, physical association | 16456542, 24012959, 25241761, (Europe PMC) | 0.70 | BioGRID, IntAct, MINT |
| PELO | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| PIGN | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| PIK3R2 | Affinity Capture-Western, Biochemical Activity | physical | 20515662, (Europe PMC) | NA | BioGRID |
| PINK1 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PKM | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| PKMYT1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| PKN2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PLEC | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| PLEKHA1 | Proximity Label-MS | physical | 27880917, (Europe PMC) | NA | BioGRID |
| PLK1 | Affinity Capture-Western, Biochemical Activity | physical | 25047839, (Europe PMC) | NA | BioGRID |
| PLPP3 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| POLDIP2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| POLM | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| PPL | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| PPP1CA | Affinity Capture-Western, anti bait coimmunoprecipitation, antibody array | physical, physical association | 17274640, (Europe PMC) | 0.52 | BioGRID, IntAct |
| PPP1R10 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| PPP3CA | Affinity Capture-Western | physical | 20605778, (Europe PMC) | NA | BioGRID |
| PRDX1 | Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | association, direct interaction, physical, physical association | 19369943, 26561776, (Europe PMC) | 0.63 | BioGRID, IntAct, MINT |
| PSMB2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| PTEN | Affinity Capture-Western, anti tag coimmunoprecipitation, bioluminescence resonance energy transfer, comigration in non denaturing gel electrophoresis, molecular sieving, pull down, x ray scattering | direct interaction, physical, physical association | 14976311, 24766807, 26299948, (Europe PMC) | 0.62, 0.63 | BioGRID, IntAct |
| PTK2 | Affinity Capture-Western, Biochemical Activity, Reconstituted Complex, proximity ligation assay | physical, physical association | 10400703, 11857088, 15717329, 23397142, 9927060, (Europe PMC) | 0.40 | BioGRID, IntAct |
| PTK2B | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PTK7 | Two-hybrid | physical | 28065597, (Europe PMC) | NA | BioGRID |
| PTPA | Affinity Capture-Western | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PTPN13 | Proximity Label-MS | physical | 27880917, (Europe PMC) | NA | BioGRID |
| PTPN14 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PXN | Affinity Capture-Western, proximity ligation assay | physical, physical association | 11857088, 23397142, (Europe PMC) | 0.40 | BioGRID, IntAct |
| QRFPR | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| RASSF2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| RCC2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RFTN1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| RHOA | Affinity Capture-Western | physical | 22720799, (Europe PMC) | NA | BioGRID |
| RNF146 | Affinity Capture-Western | physical | 25547115, (Europe PMC) | NA | BioGRID |
| RPL10A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL12 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL13 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL13A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL14 | Affinity Capture-MS, Reconstituted Complex | physical | 15717329, 26561776, (Europe PMC) | NA | BioGRID |
| RPL22L1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL23 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL27 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL27A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL31 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL38 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL7 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPLP0 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPLP2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS13 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS15A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS17 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS20 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS25 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS26 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS27 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS27A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS3 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS4X | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS6 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS9 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| S100A8 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| S100A9 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SDC1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SDC4 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SDCBP | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SETX | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SHARPIN | Affinity Capture-Western | physical | 20458142, 25152374, (Europe PMC) | NA | BioGRID |
| SKP1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SLC25A3 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SLC25A5 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SLC25A6 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| SLC36A1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| SLC9A3R1 | Affinity Capture-Western, Phenotypic Enhancement, Reconstituted Complex, filter binding, pull down, two hybrid | direct interaction, genetic, physical, physical association | 16456542, 21804599, 21990315, 23118026, 24012959, 26531778, (Europe PMC) | 0.77 | BioGRID, IntAct, MINT |
| SMTN | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| SNTB2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SOCS1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| SPOP | Affinity Capture-Western, anti tag coimmunoprecipitation, ubiquitinase assay, x-ray crystallography | direct interaction, physical, physical association, ubiquitination reaction | 24656772, 27622336, (Europe PMC) | 0.61 | BioGRID, IntAct |
| SPTAN1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SPTBN1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SSR4 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| ST8SIA6 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| STIP1 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| STUB1 | Affinity Capture-Western, Biochemical Activity, Reconstituted Complex | physical | 22427670, 26280536, (Europe PMC) | NA | BioGRID |
| SUV39H1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| THBD | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| TNKS | Affinity Capture-Western, Biochemical Activity | physical | 25547115, (Europe PMC) | NA | BioGRID |
| TNKS2 | Affinity Capture-Western, Biochemical Activity | physical | 25547115, (Europe PMC) | NA | BioGRID |
| TP53 | Affinity Capture-Western, Reconstituted Complex | physical | 12620407, 14559824, 18339852, 24141722, (Europe PMC) | NA | BioGRID |
| TRIM24 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| TRIM8 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| TTBK2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| TTC31 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| TUBA1A | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| TUBA1B | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| TUBA1C | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| TUBA3C | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| TUBB | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| TUBB2A | Affinity Capture-RNA | physical | 21532586, (Europe PMC) | NA | BioGRID |
| TUBB3 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| TUBB4B | Affinity Capture-MS, Reconstituted Complex | physical | 21532586, 26561776, (Europe PMC) | NA | BioGRID |
| TUFM | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| TXN | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| TXNDC11 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| UBAP2L | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| UBC | Affinity Capture-Western, PCA | physical | 24798731, (Europe PMC) | NA | BioGRID |
| UBE2D1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| UBE2I | Affinity Capture-Western | physical | 12620973, 22713753, (Europe PMC) | NA | BioGRID |
| UBE2L3 | Affinity Capture-Western | physical | 12620973, (Europe PMC) | NA | BioGRID |
| USP10 | Affinity Capture-MS, Affinity Capture-Western | physical | 24270891, 28852924, (Europe PMC) | NA | BioGRID |
| USP13 | Affinity Capture-MS, Affinity Capture-Western, Biochemical Activity, Co-localization, Reconstituted Complex, anti bait coimmunoprecipitation | physical, physical association | 24270891, 26280536, 26453058, (Europe PMC) | 0.40 | BioGRID, IntAct |
| USP39 | Affinity Capture-MS | physical | 24270891, (Europe PMC) | NA | BioGRID |
| USP4 | Affinity Capture-MS | physical | 24270891, (Europe PMC) | NA | BioGRID |
| USP7 | Affinity Capture-MS, Affinity Capture-Western, Biochemical Activity, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | direct interaction, physical, physical association | 18716620, 24063548, 24270891, 26280536, (Europe PMC) | 0.60 | BioGRID, IntAct |
| USP8 | Affinity Capture-MS, Affinity Capture-Western | physical | 24270891, (Europe PMC) | NA | BioGRID |
| UTP14A | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| UTRN | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| VHL | Negative Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| WASHC5 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| WNT4 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| WWP1 | Affinity Capture-MS, Affinity Capture-Western | physical | 21532586, 25547115, (Europe PMC) | NA | BioGRID |
| WWP2 | Affinity Capture-MS, Affinity Capture-Western, Biochemical Activity, Reconstituted Complex | physical | 15717329, 21532586, 22427670, 24063548, 27295432, 27852060, 28475870, (Europe PMC) | NA | BioGRID |
| XIAP | Affinity Capture-Western, Biochemical Activity | physical | 19473982, 22427670, (Europe PMC) | NA | BioGRID |
| YES1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| YME1L1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| ZNF787 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| Interacting partner | Detection method | Interaction type | PubMed IDs | Interaction Score | Source |
| ANAPC4 | Affinity Capture-MS, Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation | association, physical | 21241890, (Europe PMC) | 0.46 | BioGRID, IntAct |
| ANAPC5 | Affinity Capture-MS, Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation | association, physical | 21241890, (Europe PMC) | 0.46 | BioGRID, IntAct |
| ANAPC7 | Affinity Capture-MS, Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation | association, physical | 21241890, (Europe PMC) | 0.46 | BioGRID, IntAct |
| ANG | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| ANP32B | two hybrid array | physical association | 24412244, (Europe PMC) | 0.37 | IntAct, MINT |
| BEX1 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| BMI1 | Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, cross-linking study, fluorescence microscopy | colocalization, physical, physical association | 19903340, (Europe PMC) | 0.61 | BioGRID, IntAct, MINT |
| CAV1 | Affinity Capture-Western, coimmunoprecipitation, cosedimentation through density gradient | colocalization, physical, physical association | 12176037, (Europe PMC) | 0.50 | BioGRID, IntAct, MINT |
| CCDC180 | two hybrid array | physical association | 24412244, (Europe PMC) | 0.37 | IntAct, MINT |
| CDC27 | Affinity Capture-MS, Affinity Capture-Western, Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, fluorescence microscopy | association, colocalization, physical, physical association | 21241890, (Europe PMC) | 0.62 | BioGRID, IntAct |
| CHGB | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| COPS6 | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| CTNNB1 | Affinity Capture-Western, anti bait coimmunoprecipitation | physical, physical association | 20123964, 20372837, (Europe PMC) | 0.40 | BioGRID, IntAct |
| CXCL1 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| DBN1 | Reconstituted Complex, anti tag coimmunoprecipitation, fluorescent resonance energy transfer | physical, physical association | 23940795, 26561776, (Europe PMC) | 0.52 | BioGRID, IntAct |
| EBI-4399559 | anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, comigration in sds page, pull down, ubiquitinase assay | direct interaction, physical association, ubiquitination reaction | 18716620, 24656772, 26280536, (Europe PMC) | 0.44, 0.75 | IntAct |
| ECSCR | anti tag coimmunoprecipitation, bimolecular fluorescence complementation | physical association | 21593423, (Europe PMC) | 0.51 | IntAct |
| ENSG00000051180 | chromatin immunoprecipitation assay | association | 17218262, (Europe PMC) | 0.35 | IntAct |
| FRK | Affinity Capture-MS, Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, protein kinase assay, pull down | phosphorylation reaction, physical, physical association | 19345329, (Europe PMC) | 0.64 | BioGRID, IntAct |
| HAVCR2 | anti bait coimmunoprecipitation | association | 26492563, (Europe PMC) | 0.35 | IntAct |
| HBA1 | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| HSPA5 | cosedimentation | colocalization | 18064632, (Europe PMC) | 0.35 | IntAct |
| IL24 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| MAGI3 | Affinity Capture-Western, Reconstituted Complex, Two-hybrid, anti tag coimmunoprecipitation, pull down, two hybrid | direct interaction, physical, physical association | 10748157, (Europe PMC) | 0.63 | BioGRID, IntAct, MINT |
| MAST3 | beta galactosidase complementation, protein kinase assay, pull down | phosphorylation reaction, physical association | 15951562, (Europe PMC) | 0.59 | IntAct |
| NCOA3 | Affinity Capture-Western, anti bait coimmunoprecipitation | physical, physical association | 23514585, (Europe PMC) | 0.40 | BioGRID, IntAct |
| NEDD4 | Affinity Capture-Western, Biochemical Activity, Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | physical, physical association | 18307411, 18498243, 19345329, 21986318, 24063548, 24141722, 25047839, 25547115, 27295432, (Europe PMC) | 0.59 | BioGRID, IntAct |
| OSGIN1 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| OTUD3 | Affinity Capture-Western, Biochemical Activity, Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | physical, physical association | 26280536, (Europe PMC) | 0.59 | BioGRID, IntAct |
| PDGFRB | Co-localization, filter binding, proximity ligation assay, pull down | association, direct interaction, physical, physical association | 16456542, 24012959, 25241761, (Europe PMC) | 0.70 | BioGRID, IntAct, MINT |
| PHLPP1 | filter binding | physical association | 21804599, (Europe PMC) | 0.40 | IntAct |
| PHLPP2 | filter binding | physical association | 21804599, (Europe PMC) | 0.40 | IntAct |
| PPP1CA | Affinity Capture-Western, anti bait coimmunoprecipitation, antibody array | physical, physical association | 17274640, (Europe PMC) | 0.52 | BioGRID, IntAct |
| PRDX1 | Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | association, direct interaction, physical, physical association | 19369943, 26561776, (Europe PMC) | 0.63 | BioGRID, IntAct, MINT |
| PTEN | Affinity Capture-Western, anti tag coimmunoprecipitation, bioluminescence resonance energy transfer, comigration in non denaturing gel electrophoresis, molecular sieving, pull down, x ray scattering | direct interaction, physical, physical association | 14976311, 24766807, 26299948, (Europe PMC) | 0.62, 0.63 | BioGRID, IntAct |
| PTK2 | Affinity Capture-Western, Biochemical Activity, Reconstituted Complex, proximity ligation assay | physical, physical association | 10400703, 11857088, 15717329, 23397142, 9927060, (Europe PMC) | 0.40 | BioGRID, IntAct |
| PXN | Affinity Capture-Western, proximity ligation assay | physical, physical association | 11857088, 23397142, (Europe PMC) | 0.40 | BioGRID, IntAct |
| SLC9A3R1 | Affinity Capture-Western, Phenotypic Enhancement, Reconstituted Complex, filter binding, pull down, two hybrid | direct interaction, genetic, physical, physical association | 16456542, 21804599, 21990315, 23118026, 24012959, 26531778, (Europe PMC) | 0.77 | BioGRID, IntAct, MINT |
| SLC9A3R2 | anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, filter binding, pull down | direct interaction, physical association | 16456542, (Europe PMC) | 0.64 | IntAct, MINT |
| SPOP | Affinity Capture-Western, anti tag coimmunoprecipitation, ubiquitinase assay, x-ray crystallography | direct interaction, physical, physical association, ubiquitination reaction | 24656772, 27622336, (Europe PMC) | 0.61 | BioGRID, IntAct |
| STAT5A | genetic interference | genetic interaction | 20596030, (Europe PMC) | 0.17 | IntAct, MINT |
| TMEFF1 | two hybrid array | physical association | 24412244, (Europe PMC) | 0.37 | IntAct, MINT |
| TRMO | two hybrid array | physical association | 24412244, (Europe PMC) | 0.37 | IntAct, MINT |
| UBA1 | ubiquitinase assay | ubiquitination reaction | 24656772, (Europe PMC) | 0.44 | IntAct |
| UBE2D2 | ubiquitinase assay | ubiquitination reaction | 24656772, (Europe PMC) | 0.44 | IntAct |
| UHRF2 | antibody array | physical association | 21952639, (Europe PMC) | 0.40 | IntAct |
| USP13 | Affinity Capture-MS, Affinity Capture-Western, Biochemical Activity, Co-localization, Reconstituted Complex, anti bait coimmunoprecipitation | physical, physical association | 24270891, 26280536, 26453058, (Europe PMC) | 0.40 | BioGRID, IntAct |
| USP7 | Affinity Capture-MS, Affinity Capture-Western, Biochemical Activity, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | direct interaction, physical, physical association | 18716620, 24063548, 24270891, 26280536, (Europe PMC) | 0.60 | BioGRID, IntAct |
| UTP14A | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| Interacting partner | Detection method | Interaction type | PubMed IDs | Interaction Score | Source |
| ANP32B | two hybrid array | physical association | 24412244, (Europe PMC) | 0.37 | IntAct, MINT |
| BEX1 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| BMI1 | Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, cross-linking study, fluorescence microscopy | colocalization, physical, physical association | 19903340, (Europe PMC) | 0.61 | BioGRID, IntAct, MINT |
| CAV1 | Affinity Capture-Western, coimmunoprecipitation, cosedimentation through density gradient | colocalization, physical, physical association | 12176037, (Europe PMC) | 0.50 | BioGRID, IntAct, MINT |
| CCDC180 | two hybrid array | physical association | 24412244, (Europe PMC) | 0.37 | IntAct, MINT |
| CXCL1 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| IL24 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| MAGI3 | Affinity Capture-Western, Reconstituted Complex, Two-hybrid, anti tag coimmunoprecipitation, pull down, two hybrid | direct interaction, physical, physical association | 10748157, (Europe PMC) | 0.63 | BioGRID, IntAct, MINT |
| OSGIN1 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| PDGFRB | Co-localization, filter binding, proximity ligation assay, pull down | association, direct interaction, physical, physical association | 16456542, 24012959, 25241761, (Europe PMC) | 0.70 | BioGRID, IntAct, MINT |
| PRDX1 | Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | association, direct interaction, physical, physical association | 19369943, 26561776, (Europe PMC) | 0.63 | BioGRID, IntAct, MINT |
| SLC9A3R1 | Affinity Capture-Western, Phenotypic Enhancement, Reconstituted Complex, filter binding, pull down, two hybrid | direct interaction, genetic, physical, physical association | 16456542, 21804599, 21990315, 23118026, 24012959, 26531778, (Europe PMC) | 0.77 | BioGRID, IntAct, MINT |
| SLC9A3R2 | anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, filter binding, pull down | direct interaction, physical association | 16456542, (Europe PMC) | 0.64 | IntAct, MINT |
| STAT5A | genetic interference | genetic interaction | 20596030, (Europe PMC) | 0.17 | IntAct, MINT |
| TMEFF1 | two hybrid array | physical association | 24412244, (Europe PMC) | 0.37 | IntAct, MINT |
| TRMO | two hybrid array | physical association | 24412244, (Europe PMC) | 0.37 | IntAct, MINT |
| Interacting partner | Detection method | Interaction type | PubMed IDs | Interaction Score | Source |
| ACACA | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| ACACB | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| ADA | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| ADGRF3 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| AKAP12 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| AKT1 | Affinity Capture-Western, genetic interference | genetic interaction, physical | 20596030, 20605778, (Europe PMC) | 0.17 | BioGRID, IntAct, MINT |
| AMHR2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| AMOT | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| ANAPC4 | Affinity Capture-MS, Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation | association, physical | 21241890, (Europe PMC) | 0.46 | BioGRID, IntAct |
| ANAPC5 | Affinity Capture-MS, Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation | association, physical | 21241890, (Europe PMC) | 0.46 | BioGRID, IntAct |
| ANAPC7 | Affinity Capture-MS, Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation | association, physical | 21241890, (Europe PMC) | 0.46 | BioGRID, IntAct |
| ANG | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| ANP32B | two hybrid array | physical association | 24412244, (Europe PMC) | 0.37 | IntAct, MINT |
| ANXA2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| AR | Affinity Capture-Western, Reconstituted Complex | physical | 15205473, (Europe PMC) | NA | BioGRID |
| ARF4 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| ARHGAP26 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| ATP5F1A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| ATP5F1C | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| BAP1 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| BCAR1 | Reconstituted Complex | physical | 9927060, (Europe PMC) | NA | BioGRID |
| BCKDHA | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| BEX1 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| BLOC1S1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| BMI1 | Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, cross-linking study, fluorescence microscopy | colocalization, physical, physical association | 19903340, (Europe PMC) | 0.61 | BioGRID, IntAct, MINT |
| BRAF | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| BRD4 | Negative Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| CALM1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| CARS2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| CASP8 | Affinity Capture-MS, Affinity Capture-Western | physical | 15717329, (Europe PMC) | NA | BioGRID |
| CAV1 | Affinity Capture-Western, coimmunoprecipitation, cosedimentation through density gradient | colocalization, physical, physical association | 12176037, (Europe PMC) | 0.50 | BioGRID, IntAct, MINT |
| CBL | Affinity Capture-Western | physical | 23118026, (Europe PMC) | NA | BioGRID |
| CCDC180 | two hybrid array | physical association | 24412244, (Europe PMC) | 0.37 | IntAct, MINT |
| CCNE2 | Affinity Capture-MS, Affinity Capture-Western | physical | 15717329, (Europe PMC) | NA | BioGRID |
| CDC27 | Affinity Capture-MS, Affinity Capture-Western, Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, fluorescence microscopy | association, colocalization, physical, physical association | 21241890, (Europe PMC) | 0.62 | BioGRID, IntAct |
| CDC42EP5 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| CDH1 | Affinity Capture-Western | physical | 24811168, (Europe PMC) | NA | BioGRID |
| CDK4 | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| CDKN2A | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| CENPC | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| CFB | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| CFL1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| CHEK1 | Negative Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| CHGB | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| COPS6 | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| CRKL | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| CSNK2A1 | Affinity Capture-MS, Biochemical Activity, Reconstituted Complex | physical | 12297295, 15717329, 25047839, (Europe PMC) | NA | BioGRID |
| CSNK2A2 | Biochemical Activity, Reconstituted Complex | physical | 12297295, (Europe PMC) | NA | BioGRID |
| CTNNB1 | Affinity Capture-Western, anti bait coimmunoprecipitation | physical, physical association | 20123964, 20372837, (Europe PMC) | 0.40 | BioGRID, IntAct |
| CXCL1 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| CYB5R3 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| DBF4B | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| DBN1 | Reconstituted Complex, anti tag coimmunoprecipitation, fluorescent resonance energy transfer | physical, physical association | 23940795, 26561776, (Europe PMC) | 0.52 | BioGRID, IntAct |
| DBT | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| DCAF11 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| DDB1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| DLC1 | Affinity Capture-Western, Reconstituted Complex | physical | 19482022, (Europe PMC) | NA | BioGRID |
| DLG1 | Reconstituted Complex | physical | 11439092, (Europe PMC) | NA | BioGRID |
| DNAJA1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| DSP | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| EBI-4399559 | anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, comigration in sds page, pull down, ubiquitinase assay | direct interaction, physical association, ubiquitination reaction | 18716620, 24656772, 26280536, (Europe PMC) | 0.44, 0.75 | IntAct |
| ECSCR | anti tag coimmunoprecipitation, bimolecular fluorescence complementation | physical association | 21593423, (Europe PMC) | 0.51 | IntAct |
| EEF1A2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| EEF2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| EGFR | Affinity Capture-Western | physical | 26531778, (Europe PMC) | NA | BioGRID |
| EGR1 | Affinity Capture-Western | physical | 21354147, (Europe PMC) | NA | BioGRID |
| EIF2AK2 | Affinity Capture-Western | physical | 20029030, (Europe PMC) | NA | BioGRID |
| ELAVL1 | Affinity Capture-RNA | physical | 19322201, (Europe PMC) | NA | BioGRID |
| ELOA3 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| EME2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| ENO2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| ENSG00000051180 | chromatin immunoprecipitation assay | association | 17218262, (Europe PMC) | 0.35 | IntAct |
| EPHB1 | Affinity Capture-Western | physical | 23118026, (Europe PMC) | NA | BioGRID |
| ERBB3 | Two-hybrid | physical | 28065597, (Europe PMC) | NA | BioGRID |
| ESR1 | Reconstituted Complex | physical | 15205473, (Europe PMC) | NA | BioGRID |
| ETS1 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| FASN | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| FBN2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| FBXL6 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| FBXW7 | Affinity Capture-Western | physical | 23514585, (Europe PMC) | NA | BioGRID |
| FLNB | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| FRK | Affinity Capture-MS, Affinity Capture-Western, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, protein kinase assay, pull down | phosphorylation reaction, physical, physical association | 19345329, (Europe PMC) | 0.64 | BioGRID, IntAct |
| FZR1 | Affinity Capture-Western, Reconstituted Complex | physical | 21241890, 24811168, (Europe PMC) | NA | BioGRID |
| G3BP2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| GFRA2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| GNAI1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| GNAI2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| GNB4 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| GPC1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| GPC4 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| GPER1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| GSTM2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| HAGHL | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| HAVCR2 | anti bait coimmunoprecipitation | association | 26492563, (Europe PMC) | 0.35 | IntAct |
| HBA1 | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| HEATR6 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| HES5 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| HES6 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| HIST1H2AB | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| HNRNPL | Affinity Capture-RNA | physical | 28611215, (Europe PMC) | NA | BioGRID |
| HNRNPU | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| HSD17B1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| HSP90AA1 | Affinity Capture-MS, Positive Genetic | genetic, physical | 21532586, 28319113, (Europe PMC) | NA | BioGRID |
| HSP90AB1 | Affinity Capture-MS, Reconstituted Complex | physical | 21532586, 26561776, (Europe PMC) | NA | BioGRID |
| HSPA1A | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| HSPA1L | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| HSPA2 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| HSPA5 | cosedimentation | colocalization | 18064632, (Europe PMC) | 0.35 | IntAct |
| HSPA6 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| HSPA8 | Affinity Capture-MS, Reconstituted Complex | physical | 21532586, 26561776, (Europe PMC) | NA | BioGRID |
| HSPD1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| IFIH1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| IL24 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| INHBE | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| INS | Biochemical Activity | physical | 11439092, (Europe PMC) | NA | BioGRID |
| IRS4 | Affinity Capture-MS, Affinity Capture-Western | physical | 15717329, (Europe PMC) | NA | BioGRID |
| ITPR3 | Affinity Capture-Western | physical | 28614300, (Europe PMC) | NA | BioGRID |
| ITPRID2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| JUP | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| KAT2B | Affinity Capture-Western | physical | 16829519, (Europe PMC) | NA | BioGRID |
| KBTBD4 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| KDM6A | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| KRT14 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| LATS1 | Biochemical Activity | physical | 22641346, (Europe PMC) | NA | BioGRID |
| LDHB | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| LGALS1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| LIMA1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| LLGL2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| MAGI3 | Affinity Capture-Western, Reconstituted Complex, Two-hybrid, anti tag coimmunoprecipitation, pull down, two hybrid | direct interaction, physical, physical association | 10748157, (Europe PMC) | 0.63 | BioGRID, IntAct, MINT |
| MAP2K6 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| MAST3 | beta galactosidase complementation, protein kinase assay, pull down | phosphorylation reaction, physical association | 15951562, (Europe PMC) | 0.59 | IntAct |
| MCCC1 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| MCCC2 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| MCRS1 | Affinity Capture-Western, Far Western, Reconstituted Complex | physical | 15659546, (Europe PMC) | NA | BioGRID |
| MED12 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| MET | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| METTL26 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MPRIP | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MTHFD2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MTOR | Negative Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| MVP | Affinity Capture-Western, Reconstituted Complex, Two-hybrid | physical | 12177006, (Europe PMC) | NA | BioGRID |
| MXD1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| MYH10 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MYL12A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MYO1B | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MYO1C | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MYO1D | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| MYOF | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| NCF1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| NCOA3 | Affinity Capture-Western, anti bait coimmunoprecipitation | physical, physical association | 23514585, (Europe PMC) | 0.40 | BioGRID, IntAct |
| NDFIP1 | Affinity Capture-Western, PCA | physical | 20534535, 22213801, 24798731, (Europe PMC) | NA | BioGRID |
| NDFIP2 | Affinity Capture-Western | physical | 20534535, (Europe PMC) | NA | BioGRID |
| NEDD4 | Affinity Capture-Western, Biochemical Activity, Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | physical, physical association | 18307411, 18498243, 19345329, 21986318, 24063548, 24141722, 25047839, 25547115, 27295432, (Europe PMC) | 0.59 | BioGRID, IntAct |
| NF1 | Positive Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| NGB | Reconstituted Complex | physical | 23904011, (Europe PMC) | NA | BioGRID |
| NME1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| NOTCH1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| OPRL1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| OSBP2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| OSGIN1 | Two-hybrid, two hybrid array | physical, physical association | 25640309, (Europe PMC) | 0.37 | BioGRID, IntAct, MINT |
| OTUD3 | Affinity Capture-Western, Biochemical Activity, Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | physical, physical association | 26280536, (Europe PMC) | 0.59 | BioGRID, IntAct |
| P3H4 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PARK7 | Affinity Capture-Western, Reconstituted Complex | physical | 19885556, 21426932, (Europe PMC) | NA | BioGRID |
| PBK | Affinity Capture-Western, Biochemical Activity | physical | 24012691, (Europe PMC) | NA | BioGRID |
| PBX3 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| PC | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| PCCA | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| PCCB | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| PDGFRA | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PDGFRB | Co-localization, filter binding, proximity ligation assay, pull down | association, direct interaction, physical, physical association | 16456542, 24012959, 25241761, (Europe PMC) | 0.70 | BioGRID, IntAct, MINT |
| PELO | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| PHLPP1 | filter binding | physical association | 21804599, (Europe PMC) | 0.40 | IntAct |
| PHLPP2 | filter binding | physical association | 21804599, (Europe PMC) | 0.40 | IntAct |
| PIGN | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| PIK3R2 | Affinity Capture-Western, Biochemical Activity | physical | 20515662, (Europe PMC) | NA | BioGRID |
| PINK1 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PKM | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| PKMYT1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| PKN2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PLEC | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| PLEKHA1 | Proximity Label-MS | physical | 27880917, (Europe PMC) | NA | BioGRID |
| PLK1 | Affinity Capture-Western, Biochemical Activity | physical | 25047839, (Europe PMC) | NA | BioGRID |
| PLPP3 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| POLDIP2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| POLM | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| PPL | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| PPP1CA | Affinity Capture-Western, anti bait coimmunoprecipitation, antibody array | physical, physical association | 17274640, (Europe PMC) | 0.52 | BioGRID, IntAct |
| PPP1R10 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| PPP3CA | Affinity Capture-Western | physical | 20605778, (Europe PMC) | NA | BioGRID |
| PRDX1 | Reconstituted Complex, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | association, direct interaction, physical, physical association | 19369943, 26561776, (Europe PMC) | 0.63 | BioGRID, IntAct, MINT |
| PSMB2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| PTEN | Affinity Capture-Western, anti tag coimmunoprecipitation, bioluminescence resonance energy transfer, comigration in non denaturing gel electrophoresis, molecular sieving, pull down, x ray scattering | direct interaction, physical, physical association | 14976311, 24766807, 26299948, (Europe PMC) | 0.62, 0.63 | BioGRID, IntAct |
| PTK2 | Affinity Capture-Western, Biochemical Activity, Reconstituted Complex, proximity ligation assay | physical, physical association | 10400703, 11857088, 15717329, 23397142, 9927060, (Europe PMC) | 0.40 | BioGRID, IntAct |
| PTK2B | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PTK7 | Two-hybrid | physical | 28065597, (Europe PMC) | NA | BioGRID |
| PTPA | Affinity Capture-Western | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PTPN13 | Proximity Label-MS | physical | 27880917, (Europe PMC) | NA | BioGRID |
| PTPN14 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| PXN | Affinity Capture-Western, proximity ligation assay | physical, physical association | 11857088, 23397142, (Europe PMC) | 0.40 | BioGRID, IntAct |
| QRFPR | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| RASSF2 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| RCC2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RFTN1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| RHOA | Affinity Capture-Western | physical | 22720799, (Europe PMC) | NA | BioGRID |
| RNF146 | Affinity Capture-Western | physical | 25547115, (Europe PMC) | NA | BioGRID |
| RPL10A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL12 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL13 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL13A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL14 | Affinity Capture-MS, Reconstituted Complex | physical | 15717329, 26561776, (Europe PMC) | NA | BioGRID |
| RPL22L1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL23 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL27 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL27A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL31 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL38 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPL7 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPLP0 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPLP2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS13 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS15A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS17 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS20 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS25 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS26 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS27 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS27A | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS3 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS4X | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS6 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| RPS9 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| S100A8 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| S100A9 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SDC1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SDC4 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SDCBP | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SETX | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SHARPIN | Affinity Capture-Western | physical | 20458142, 25152374, (Europe PMC) | NA | BioGRID |
| SKP1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SLC25A3 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SLC25A5 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SLC25A6 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| SLC36A1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| SLC9A3R1 | Affinity Capture-Western, Phenotypic Enhancement, Reconstituted Complex, filter binding, pull down, two hybrid | direct interaction, genetic, physical, physical association | 16456542, 21804599, 21990315, 23118026, 24012959, 26531778, (Europe PMC) | 0.77 | BioGRID, IntAct, MINT |
| SLC9A3R2 | anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, filter binding, pull down | direct interaction, physical association | 16456542, (Europe PMC) | 0.64 | IntAct, MINT |
| SMTN | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| SNTB2 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SOCS1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| SPOP | Affinity Capture-Western, anti tag coimmunoprecipitation, ubiquitinase assay, x-ray crystallography | direct interaction, physical, physical association, ubiquitination reaction | 24656772, 27622336, (Europe PMC) | 0.61 | BioGRID, IntAct |
| SPTAN1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SPTBN1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| SSR4 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| ST8SIA6 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| STAT5A | genetic interference | genetic interaction | 20596030, (Europe PMC) | 0.17 | IntAct, MINT |
| STIP1 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| STUB1 | Affinity Capture-Western, Biochemical Activity, Reconstituted Complex | physical | 22427670, 26280536, (Europe PMC) | NA | BioGRID |
| SUV39H1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| THBD | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| TMEFF1 | two hybrid array | physical association | 24412244, (Europe PMC) | 0.37 | IntAct, MINT |
| TNKS | Affinity Capture-Western, Biochemical Activity | physical | 25547115, (Europe PMC) | NA | BioGRID |
| TNKS2 | Affinity Capture-Western, Biochemical Activity | physical | 25547115, (Europe PMC) | NA | BioGRID |
| TP53 | Affinity Capture-Western, Reconstituted Complex | physical | 12620407, 14559824, 18339852, 24141722, (Europe PMC) | NA | BioGRID |
| TRIM24 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| TRIM8 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| TRMO | two hybrid array | physical association | 24412244, (Europe PMC) | 0.37 | IntAct, MINT |
| TTBK2 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| TTC31 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| TUBA1A | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| TUBA1B | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| TUBA1C | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| TUBA3C | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| TUBB | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| TUBB2A | Affinity Capture-RNA | physical | 21532586, (Europe PMC) | NA | BioGRID |
| TUBB3 | Affinity Capture-MS | physical | 21532586, (Europe PMC) | NA | BioGRID |
| TUBB4B | Affinity Capture-MS, Reconstituted Complex | physical | 21532586, 26561776, (Europe PMC) | NA | BioGRID |
| TUFM | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| TXN | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| TXNDC11 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| UBA1 | ubiquitinase assay | ubiquitination reaction | 24656772, (Europe PMC) | 0.44 | IntAct |
| UBAP2L | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| UBC | Affinity Capture-Western, PCA | physical | 24798731, (Europe PMC) | NA | BioGRID |
| UBE2D1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| UBE2D2 | ubiquitinase assay | ubiquitination reaction | 24656772, (Europe PMC) | 0.44 | IntAct |
| UBE2I | Affinity Capture-Western | physical | 12620973, 22713753, (Europe PMC) | NA | BioGRID |
| UBE2L3 | Affinity Capture-Western | physical | 12620973, (Europe PMC) | NA | BioGRID |
| UHRF2 | antibody array | physical association | 21952639, (Europe PMC) | 0.40 | IntAct |
| USP10 | Affinity Capture-MS, Affinity Capture-Western | physical | 24270891, 28852924, (Europe PMC) | NA | BioGRID |
| USP13 | Affinity Capture-MS, Affinity Capture-Western, Biochemical Activity, Co-localization, Reconstituted Complex, anti bait coimmunoprecipitation | physical, physical association | 24270891, 26280536, 26453058, (Europe PMC) | 0.40 | BioGRID, IntAct |
| USP39 | Affinity Capture-MS | physical | 24270891, (Europe PMC) | NA | BioGRID |
| USP4 | Affinity Capture-MS | physical | 24270891, (Europe PMC) | NA | BioGRID |
| USP7 | Affinity Capture-MS, Affinity Capture-Western, Biochemical Activity, anti bait coimmunoprecipitation, anti tag coimmunoprecipitation, pull down | direct interaction, physical, physical association | 18716620, 24063548, 24270891, 26280536, (Europe PMC) | 0.60 | BioGRID, IntAct |
| USP8 | Affinity Capture-MS, Affinity Capture-Western | physical | 24270891, (Europe PMC) | NA | BioGRID |
| UTP14A | Two-hybrid, two hybrid pooling approach | physical, physical association | 16169070, (Europe PMC) | 0.37 | BioGRID, IntAct |
| UTRN | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| VHL | Negative Genetic | genetic | 28319113, (Europe PMC) | NA | BioGRID |
| WASHC5 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| WNT4 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |
| WWP1 | Affinity Capture-MS, Affinity Capture-Western | physical | 21532586, 25547115, (Europe PMC) | NA | BioGRID |
| WWP2 | Affinity Capture-MS, Affinity Capture-Western, Biochemical Activity, Reconstituted Complex | physical | 15717329, 21532586, 22427670, 24063548, 27295432, 27852060, 28475870, (Europe PMC) | NA | BioGRID |
| XIAP | Affinity Capture-Western, Biochemical Activity | physical | 19473982, 22427670, (Europe PMC) | NA | BioGRID |
| YES1 | Reconstituted Complex | physical | 26561776, (Europe PMC) | NA | BioGRID |
| YME1L1 | Synthetic Growth Defect | genetic | 24104479, (Europe PMC) | NA | BioGRID |
| ZNF787 | Affinity Capture-MS | physical | 15717329, (Europe PMC) | NA | BioGRID |