241 human active and 13 inactive phosphatases in total;
194 phosphatases have substrate data;
--------------------------------
336 protein substrates;
83 non-protein substrates;
1215 dephosphorylation interactions;
--------------------------------
299 KEGG pathways;
876 Reactome pathways;
--------------------------------
last scientific update: 11 Mar, 2019
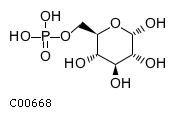
Hydrolyzes glucose-6-phosphate to glucose in theendoplasmic reticulum May form with the glucose-6-phosphatetransporter (SLC37A4/G6PT) a ubiquitously expressed complexresponsible for glucose production through glycogenolysis andgluconeogenesis Probably required for normal neutrophil function
Glycolysis is the process of converting glucose into pyruvate and generating small amounts of ATP (energy) and NADH (reducing power). It is a central pathway that produces important precursor metabolites: six-carbon compounds of glucose-6P and fructose-6P and three-carbon compounds of glycerone-P, glyceraldehyde-3P, glycerate-3P, phosphoenolpyruvate, and pyruvate [MD:M00001]. Acetyl-CoA, another important precursor metabolite, is produced by oxidative decarboxylation of pyruvate [MD:M00307]. When the enzyme genes of this pathway are examined in completely sequenced genomes, the reaction steps of three-carbon compounds from glycerone-P to pyruvate form a conserved core module [MD:M00002], which is found in almost all organisms and which sometimes contains operon structures in bacterial genomes. Gluconeogenesis is a synthesis pathway of glucose from noncarbohydrate precursors. It is essentially a reversal of glycolysis with minor variations of alternative paths [MD:M00003].
The forkhead box O (FOXO) family of transcription factors regulates the expression of genes in cellular physiological events including apoptosis, cell-cycle control, glucose metabolism, oxidative stress resistance, and longevity. A central regulatory mechanism of FOXO proteins is phosphorylation by the serine-threonine kinase Akt/protein kinase B (Akt/PKB), downstream of phosphatidylinositol 3-kinase (PI3K), in response to insulin or several growth factors. Phosphorylation at three conserved residues results in the export of FOXO proteins from the nucleus to the cytoplasm, thereby decreasing expression of FOXO target genes. In contrast, the stress-activated c-Jun N-terminal kinase (JNK) and the energy sensing AMP-activated protein kinase (AMPK), upon oxidative and nutrient stress stimuli phosphorylate and activate FoxOs. Aside from PKB, JNK and AMPK, FOXOs are regulated by multiple players through several post-translational modifications, including phosphorylation, but also acetylation, methylation and ubiquitylation.
The phosphatidylinositol 3' -kinase(PI3K)-Akt signaling pathway is activated by many types of cellular stimuli or toxic insults and regulates fundamental cellular functions such as transcription, translation, proliferation, growth, and survival. The binding of growth factors to their receptor tyrosine kinase (RTK) or G protein-coupled receptors (GPCR) stimulates class Ia and Ib PI3K isoforms, respectively. PI3K catalyzes the production of phosphatidylinositol-3,4,5-triphosphate (PIP3) at the cell membrane. PIP3 in turn serves as a second messenger that helps to activate Akt. Once active, Akt can control key cellular processes by phosphorylating substrates involved in apoptosis, protein synthesis, metabolism, and cell cycle.
AMP-activated protein kinase (AMPK) is a serine threonine kinase that is highly conserved through evolution. AMPK system acts as a sensor of cellular energy status. It is activated by increases in the cellular AMP:ATP ratio caused by metabolic stresses that either interfere with ATP production (eg, deprivation for glucose or oxygen) or that accelerate ATP consumption (eg, muscle contraction). Several upstream kinases, including liver kinase B1 (LKB1), calcium/calmodulin kinase kinase-beta (CaMKK beta), and TGF-beta-activated kinase-1 (TAK-1), can activate AMPK by phosphorylating a threonine residue on its catalytic alpha-subunit. Once activated, AMPK leads to a concomitant inhibition of energy-consuming biosynthetic pathways, such as protein, fatty acid and glycogen synthesis, and activation of ATP-producing catabolic pathways, such as fatty acid oxidation and glycolysis.
Insulin binding to its receptor results in the tyrosine phosphorylation of insulin receptor substrates (IRS) by the insulin receptor tyrosine kinase (INSR). This allows association of IRSs with the regulatory subunit of phosphoinositide 3-kinase (PI3K). PI3K activates 3-phosphoinositide-dependent protein kinase 1 (PDK1), which activates Akt, a serine kinase. Akt in turn deactivates glycogen synthase kinase 3 (GSK-3), leading to activation of glycogen synthase (GYS) and thus glycogen synthesis. Activation of Akt also results in the translocation of GLUT4 vesicles from their intracellular pool to the plasma membrane, where they allow uptake of glucose into the cell. Akt also leads to mTOR-mediated activation of protein synthesis by eIF4 and p70S6K. The translocation of GLUT4 protein is also elicited through the CAP/Cbl/TC10 pathway, once Cbl is phosphorylated by INSR.Other signal transduction proteins interact with IRS including GRB2. GRB2 is part of the cascade including SOS, RAS, RAF and MEK that leads to activation of mitogen-activated protein kinase (MAPK) and mitogenic responses in the form of gene transcription. SHC is another substrate of INSR. When tyrosine phosphorylated, SHC associates with GRB2 and can thus activate the RAS/MAPK pathway independently of IRS-1.
Increased adipocyte volume and number are positively correlated with leptin production, and negatively correlated with production of adiponectin.Leptin is an important regulator of energy intake and metabolic rate primarily by acting at hypothalamic nuclei. Leptin exerts its anorectic effects by modulating the levels of neuropeptides such as NPY, AGRP, and alpha-MSH. This leptin action is through the JAK kinase, STAT3 phosphorylation, and nuclear transcriptional effect.Adiponectin lowers plasma glucose and FFAs. These effects are partly accounted for by adiponectin-induced AMPK activation, which in turn stimulates skeletal muscle fatty acid oxidation and glucose uptake. Furthermore, activation of AMPK by adiponectin suppresses endogenous glucose production, concomitantly with inhibition of PEPCK and G6Pase expression.The proinflammatory cytokine TNFalpha has been implicated as a link between obesity and insulin resistance. TNFalpha interferes with early steps of insulin signaling. Several data have shown that TNFalpha inhibits IRS1 tyrosine phosphorylation by promoting its serine phosphorylation. Among the serine/threonine kinases activated by TNFalpha, JNK, mTOR and IKK have been shown to be involved in this phosphorylation.
Glucagon is conventionally regarded as a counterregulatory hormone for insulin and plays a critical anti-hypoglycemic role by maintaining glucose homeostasis in both animals and humans. To increase blood glucose, glucagon promotes hepatic glucose output by increasing glycogenolysis and gluconeogenesis and by decreasing glycogenesis and glycolysis in a concerted fashion via multiple mechanisms. Glucagon also stimulates hepatic mitochondrial beta-oxidation to supply energy for glucose production. Glucagon performs its main effect via activation of adenylate cyclase. The adenylate-cyclase-derived cAMP activates protein kinase A (PKA), which then phosphorylates downstream targets, such as cAMP response element binding protein (CREB) and the bifunctional enzyme 6-phosphofructo-2-kinase/ fructose-2,6-bisphosphatase (one of the isoforms being PFK/FBPase 1, encoded by PFKFB1).
Insulin resistance is a condition where cells become resistant to the effects of insulin. It is often found in people with health disorders, including obesity, type 2 diabetes mellitus, non-alcoholic fatty liver disease, and cardiovascular diseases. In this diagram multiple mechanisms underlying insulin resistance are shown: (a) increased phosphorylation of IRS (insulin receptor substrate) protein through serine/threonine kinases, such as JNK1 and IKKB, and protein kinase C, (b) increased IRS-1 proteasome degradation via mTOR signaling pathway, (c) decreased activation of signaling molecules including PI3K and AKT, (d) increase in activity of phosphatases including PTPs, PTEN, and PP2A. Regulatory actions such as oxidative stress, mitochondrial dysfunction, accumulation of intracellular lipid derivatives (diacylglycrol and ceramides), and inflammation (via IL-6 and TNFA) contribute to these mechanisms. Consequently, insulin resistance causes reduced GLUT4 translocation, resulting in glucose takeup and glycogen synthesis in skeletal muscle as well as increased hepatic gluconeogenesis and decreased glycogen synthesis in liver. At the bottom of the diagram, interplay between O-GlcNAcylation and serine/threonine phosphorylation is shown. Studies suggested that elevated O-GlcNAc level was correlated to high glucose-induced insulin resistance. Donor UDP-GlcNAc is induced through hexosamine biosynthesis pathway and added to proteins by O-GlcNAc transferase. Elevation of O-GlcNAc modification alters phosphorylation and function of key insulin signaling proteins including IRS-1, PI3K, PDK1, Akt and other transcription factor and cofactors, resulting in the attenuation of insulin signaling cascade.
Dietary carbohydrate in humans and omnivorous animals is a major nutrient. The carbohydrates that we ingest vary from the lactose in milk to complex carbohydrates. These carbohydrates are digested to monosaccharides, mostly glucose, galactose and fructose, prior to absorption in the small intestine. Glucose and galactose are initially transported into the enterocyte by SGLT1 located in the apical brush border membrane and then exit through the basolateral membrane by either GLUT2 or exocytosis. In a new model of intestinal glucose absorption, transport by SGLT1 induces rapid insertion and activation of GLUT2 in the brush border membrane by a PKC betaII-dependent mechanism. Moreover, trafficking of apical GLUT2 is rapidly up-regulated by glucose and artificial sweeteners, which act through T1R2 + T1R3/alpha-gustducin to activate PLC-beta2 and PKC-beta II. Fructose is transported separately by the brush border GLUT5 and then released out of the enterocyte into the blood by GLUT2.
Glucose-6-phosphatase 3 (G6PC3) associated with the endoplasmic reticulum membrane normally catalyzes the hydrolysis of glucose-6-phosphate to glucose and orthophosphate. In the body, this enzyme is ubiquitously expressed; mutations that inactivate it are associated with severe congenital neutropenia (but not with fasting hypoglycemia or lactic acidemia) (Boztug et al. 2009, 2012)
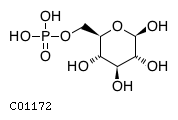
The reactions of gluconeogenesis convert mitochondrial pyruvate to cytosolic glucose 6-phosphate which in turn can be hydrolyzed to glucose and exported from the cell. Gluconeogenesis is confined to cells of the liver and kidney and enables glucose synthesis from molecules such as lactate and alanine and other amino acids when exogenous glucose is not available (reviewed, e.g., by Gerich 1993). The process of gluconeogenesis as diagrammed below occurs in two parts: a network of reactions converts mitochondrial pyruvate to cytosolic phosphoenolpyruvate; then phosphoenolpyruvate is converted to glucose 6-phosphate in a single sequence of cytosolic reactions.
Three variants of the first part of the process are physiologically important. 1) A series of transport and transamination reactions convert mitochondrial oxaloacetate to cytosolic oxaloacetate which is converted to phosphoenolpyruvate by a hormonally regulated, cytosolic isoform of phosphoenolpyruvate carboxykinase. This variant allows regulated glucose synthesis from lactate. 2) Mitochondrial oxaloacetate is reduced to malate, which is exported to the cytosol and re-oxidized to oxaloacetate. This variant provides reducing equivalents to the cytosol, needed for glucose synthesis from amino acids such as alanine and glutamine. 3) Constitutively expressed mitochondrial phosphoenolpyruvate carboxykinase catalyzes the conversion of mitochondrial oxaloacetate to phosphoenolpyruvate which is then transported to the cytosol. The exact path followed by any one molecule of pyruvate through this reaction network is determined by the tissue in which the reactions are occurring, the source of the pyruvate, and the physiological stress that triggered gluconeogenesis.
In all cases, the synthesis of glucose from two molecules of pyruvate requires the generation and consumption of two reducing equivalents as cytosolic NADH + H+. For pyruvate derived from lactate (variants 1 and 3), NADH + H+ is generated with the oxidation of lactate to pyruvate in the cytosol (a reaction of pyruvate metabolism not shown in the diagram). For pyruvate derived from amino acids (variant 2), mitochondrial NADH + H+ generated by glutamate dehydrogenase (a reaction of amino acid metabolism, not shown) is used to reduce oxaloacetate to malate, which is transported to the cytosol and re-oxidized, generating cytosolic NADH + H+. The synthesis of glucose from pyruvate also requires the consumption of six high-energy phosphates, four from ATP and two from GTP.
In the second part of gluconeogenesis, cytosolic phosphoenolpyruvate, however derived, is converted to fructose 1,6-bisphosphate by reactions that are the reverse of steps of glycolysis. Hydrolysis of fructose 1,6-bisphosphate to fructose 6-phosphate is catalyzed by fructose 1,6-bisphosphatase, and fructose 6-phosphate is reversibly isomerized to glucose 6-phosphate

 Statistics
Statistics