Statistics
Statistics
241 human active and 13 inactive phosphatases in total;
194 phosphatases have substrate data;
--------------------------------
336 protein substrates;
83 non-protein substrates;
1215 dephosphorylation interactions;
--------------------------------
299 KEGG pathways;
876 Reactome pathways;
--------------------------------
last scientific update:
11 Mar, 2019
194 phosphatases have substrate data;
--------------------------------
336 protein substrates;
83 non-protein substrates;
1215 dephosphorylation interactions;
--------------------------------
299 KEGG pathways;
876 Reactome pathways;
--------------------------------
last scientific update:
11 Mar, 2019
Top
TIGAR
| Gene Name | TIGAR (QuickGO) | (A quick tutorial to explore the interctive visulaization) |
| Synonyms | TIGAR , C12orf5 | |
| Protein Name | TIGAR |
|
| Alternative Name(s) |
Fructose-2,6-bisphosphatase TIGAR;3.1.3.46;TP53-induced glycolysis and apoptosis regulator;TP53-induced glycolysis regulatory phosphatase; | |
| EntrezGene ID | 57103 (Comparitive Toxicogenomics) | |
| UniProt AC (Human) | Q9NQ88 (protein sequence) | |
| Enzyme Class | EC 3.1.3.46 (BRENDA | |
| Molecular Weight | 30063 Dalton | |
| Protein Length | 270 amino acids (AA) | |
| Genome Browsers | NCBI | ENSG00000078237 (Ensembl) | UCSC | |
| Crosslinking annotations | Query our ID-mapping table | |
| Orthologues | Quest For Orthologues (QFO) | GeneTree | |
| Classification | Superfamily: histidine phosphatase (HP) | Historic class: Phosphoglycerate mutase | CATH ID: 3.40.50.1240 | SCOP Fold: HP | |
| Phosphatase activity | active | Catalytic signature motif: unknown | |
| Phosphorylation Network | Visualize | |
| Domain organization, Expression, Diseases | (show / hide) |
| Protein Domain | InterPro | Pfam | SMART | 3D Structure | PDB | DrugPort | ModBase | SwissModel |
| Polypeptide organization (Pfam) | No Pfam domain found | ||
| Catalytic Site | Mechanism and Catalytic Site Atlas | ||
| Gene Expression | Gene Expression Atlas | Diseases | OMIM | DisGeNet | ClinVar |
| Protein-protein interactions | STRING | IntAct | MINT | BioGRID | ||
| Phosphosites | Phospho.ELM | PhosphoSitePlus | ||
| Localization, Function, Catalytic activity and Sequence | (show / hide) |
| Localization (UniProt annotation) | |||
| Cytoplasm Nucleus Mitochondrion Note=Translocated to themitochondria during hypoxia in a HIF1A-dependent manner(PubMed:23185017) Colocalizes with HK2 in the mitochondria duringhypoxia (PubMed:23185017) Translocated to the nucleus duringhypoxia and/or genome stress-induced DNA damage responses incancer cells (PubMed:25928429) Translocation to the mitochondriais enhanced in ischemic cortex after reperfusion and/or duringoxygen and glucose deprivation (OGD)/reoxygenation insult inprimary neurons (By similarity) | |||
| Function (UniProt annotation) | |||
| Fructose-bisphosphatase hydrolyzing fructose-2,6-bisphosphate as well as fructose-1,6-bisphosphate(PubMed:19015259) Acts as a negative regulator of glycolysis bylowering intracellular levels of fructose-2,6-bisphosphate in ap53/TP53-dependent manner, resulting in the pentose phosphatepathway (PPP) activation and NADPH production (PubMed:16839880,PubMed:22887998) Contributes to the generation of reducedglutathione to cause a decrease in intracellular reactive oxygenspecies (ROS) content, correlating with its ability to protectcells from oxidative or metabolic stress-induced cell death(PubMed:16839880, PubMed:19713938, PubMed:23726973,PubMed:22887998, PubMed:23817040) Plays a role in promotingprotection against cell death during hypoxia by decreasingmitochondria ROS levels in a HK2-dependent manner through amechanism that is independent of its fructose-bisphosphataseactivity (PubMed:23185017) In response to cardiac damage stress,mediates p53-induced inhibition of myocyte mitophagy through ROSlevels reduction and the subsequent inactivation of BNIP3 Reducedmitophagy results in an enhanced apoptotic myocyte cell death, andexacerbates cardiac damage (By similarity) Plays a role in adultintestinal regeneration; contributes to the growth, proliferationand survival of intestinal crypts following tissue ablation(PubMed:23726973) Plays a neuroprotective role against ischemicbrain damage by enhancing PPP flux and preserving mitochondriafunctions (By similarity) Protects glioma cells from hypoxia- andROS-induced cell death by inhibiting glycolysis and activatingmitochondrial energy metabolism and oxygen consumption in a TKTL1-dependent and p53/TP53-independent manner (PubMed:22887998) Playsa role in cancer cell survival by promoting DNA repair throughactivating PPP flux in a CDK5-ATM-dependent signaling pathwayduring hypoxia and/or genome stress-induced DNA damage responses(PubMed:25928429) Involved in intestinal tumor progression(PubMed:23726973) | |||
| Catalytic Activity (UniProt annotation) | |||
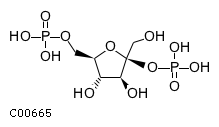
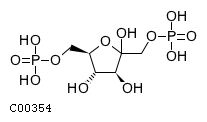
| Beta-D-fructose 2,6-bisphosphate + H(2)O = D-fructose 6-phosphate + phosphate | |||
| Protein Sequence | |||
| MARFALTVVRHGETRFNKEKIIQGQGVDEPLSETGFKQAAAAGIFLNNVKFTHAFSSDLMRTKQTMHGILERSKFCKDMT VKYDSRLRERKYGVVEGKALSELRAMAKAAREECPVFTPPGGETLDQVKMRGIDFFEFLCQLILKEADQKEQFSQGSPSN CLETSLAEIFPLGKNHSSKVNSDSGIPGLAASVLVVSHGAYMRSLFDYFLTDLKCSLPATLSRSELMSVTPNTGMSLFII NFEEGREVKPTVQCICMNLQDHLNGLTETR |
| Motif information from Eukaryotic Linear Motif atlas (ELM) | (show / hide) |
| ELM Motif | Motif Instance | Description | Motif Regex |
| NA | NA | NA | NA |
| Gene Ontology (P: Process; F: Function and C: Component terms) | (show / hide) | |
| GO:0002931 | P:response to ischemia |
| GO:0004083 | F:bisphosphoglycerate 2-phosphatase activity |
| GO:0004331 | F:fructose-2,6-bisphosphate 2-phosphatase activity |
| GO:0005622 | C:intracellular |
| GO:0005634 | C:nucleus |
| GO:0005737 | C:cytoplasm |
| GO:0005741 | C:mitochondrial outer membrane |
| GO:0005829 | C:cytosol |
| GO:0006003 | P:fructose 2,6-bisphosphate metabolic process |
| GO:0006914 | P:autophagy |
| GO:0006915 | P:apoptotic process |
| GO:0006974 | P:cellular response to DNA damage stimulus |
| GO:0009410 | P:response to xenobiotic stimulus |
| GO:0010332 | P:response to gamma radiation |
| GO:0010666 | P:positive regulation of cardiac muscle cell apoptotic process |
| GO:0030388 | P:fructose 1,6-bisphosphate metabolic process |
| GO:0043069 | P:negative regulation of programmed cell death |
| GO:0043456 | P:regulation of pentose-phosphate shunt |
| GO:0045739 | P:positive regulation of DNA repair |
| GO:0045820 | P:negative regulation of glycolytic process |
| GO:0060576 | P:intestinal epithelial cell development |
| GO:0071279 | P:cellular response to cobalt ion |
| GO:0071456 | P:cellular response to hypoxia |
| GO:1901215 | P:negative regulation of neuron death |
| GO:1901525 | P:negative regulation of mitophagy |
| GO:1902153 | P:regulation of response to DNA damage checkpoint signaling |
| GO:1903301 | P:positive regulation of hexokinase activity |
| GO:1904024 | P:negative regulation of glucose catabolic process to lactate via pyruvate |
| GO:2000378 | P:negative regulation of reactive oxygen species metabolic process |