241 human active and 13 inactive phosphatases in total;
194 phosphatases have substrate data;
--------------------------------
336 protein substrates;
83 non-protein substrates;
1215 dephosphorylation interactions;
--------------------------------
299 KEGG pathways;
876 Reactome pathways;
--------------------------------
last scientific update: 11 Mar, 2019
Autophagy (or macroautophagy) is a cellular catabolic pathway involving in protein degradation, organelle turnover, and non-selective breakdown of cytoplasmic components, which is evolutionarily conserved among eukaryotes and exquisitely regulated. This progress initiates with production of the autophagosome, a double-membrane intracellular structure of reticular origin that engulfs cytoplasmic contents and ultimately fuses with lysosomes for cargo degradation. Autophagy is regulated in response to extra- or intracellular stress and signals such as starvation, growth factor deprivation and ER stress. Constitutive level of autophagy plays an important role in cellular homeostasis and maintains quality control of essential cellular components.
Macroautophagy (hereafter referred to as autophagy) acts as a buffer against starvation by liberating building materials and energy sources from cellular components. It has additional roles in embryonic development, removal of apoptotic cells or organelles, antigen presentation, protection against toxins and as a degradation route for aggregate-prone proteins and infectious agents. The dysregulation of autophagy is involved in several human diseases, for example, Crohn's disease, cancer and neurodegeneration (Ravikumar et al. 2010).Autophagy is highly conserved from yeast to humans; much of the machinery was first identified in yeast (see Klionsky et al. 2011). Initially, double-membraned cup-shaped structures called the isolation membrane or phagophore engulf portions of cytoplasm. The membranes fuse to form the autophagosome. In yeast cells, autophagosomes are formed at the phagophore assembly site (PAS) next to the vacuole. In mammals, autophagosomes appear throughout the cytoplasm then move along microtubules towards the microtubule-organising centre. This transport requires microtubules and the function of dynein motor proteins; depolymerization of microtubules or inhibition of dynein-dependent transport results in inhibition of autophagy (Kochl et al. 2006, Kimura et al. 2008). Autophagosomes fuse with lysosomes forming autolysosomes whose contents are degraded by lysosomal hydrolases (Mizushima et al. 2011).The origins of the autophagosomal membrane and the incorporation of existing membrane material have been extensively debated. The endoplasmic reticulum (ER), mitochondria, mitochondria-associated ER membranes (MAMs), the Golgi, the plasma membrane and recycling endosomes have all been implicated in the nucleation of the isolation membrane and subsequent growth of the membrane (Lamb et al. 2013). Recently 3D tomographic imaging of isolation membranes has shown the cup-shaped isolation membrane tightly sandwiched between two sheets of ER and physically connected to the ER through a narrow membrane tube (Hayashi-Nishino et al. 2009, Yla-Anttila et al. 2009). This suggests that isolation membrane formation and elongation are guided by adjacent ER sheets, supporting the now prevalent 'ER cradle' model, which suggests that the isolation membrane arises from the ER (Hayashi-Nishino et al. 2009, Shibutani & Yoshimori 2014).Autophagy is tightly regulated. The induction of autophagy in response to starvation is partly mediated by inactivation of the mammalian target of rapamycin (mTOR) (Noda & Ohsumi 1998) and activation of Jun N-terminal kinase (JNK), while energy loss induces autophagy by activation of AMP kinase (AMPK). Other pathways regulating autophagy are regulated by calcium, cyclic AMP, calpains and the inositol trisphosphate (IP3) receptor (Rubinsztein et al. 2012). In mammals, two complexes cooperatively produce the isolation membrane. The ULK complex consists of ULK1/2, ATG13, (FIP200) and ATG101 (Alers et al. 2012). The PIK3C3-containing Beclin-1 complex consists of PIK3C3 (Vps34), BECN1 (Beclin-1, Atg6), PIK3R4 (p150, Vps15) and ATG14 (Barkor) (Matsunaga et al. 2009, Zhong et al. 2009). A similar complex where ATG14 is replaced by UVRAG functions later in autophagosome maturation and endocytic traffic (Itakura et al. 2008, Liang et al. 2008). Binding of KIAA0226 to this complex negatively regulates the maturation process (Matsunaga et al. 2009). The ULK and Beclin-1 complexes are recruited to specific autophagosome nucleation regions where they stimulate phosphatidylinositol-3-phosphate (PI3P) production and facilitate the elongation and initial membrane curvature of the phagophore membrane (Carlsson & Simonsen 2015).The ULK complex is considered the most upstream component of the mammalian autophagy pathway (Itakura & Mizushima 2010), acting as an integrator of the autophagy signals downstream of mTORC1. It is not fully understood how ULK1 is modulated in response to environmental cues. Phosphorylation plays an essential role (Dunlop & Tee 2013) but it is not clear how phosphorylation regulates ULK1 activities (Ravikumar et al. 2010). ULK1 kinase activity is required for autophagy, but the substrate(s) of ULK1 that mediate its autophagic function are not certain. ULK1 may also have kinase-independent functions in autophagy (Wong et al. 2013). PIK3C3 (Vps34) is a class III phosphatidylinositol 3-kinase that produces PI3P. It is essential for the early stages of autophagy and colocalizes strongly with early autophagosome markers (Axe et al. 2008). BECN1 binds several further proteins that affect autophagosome formation. Partners that induce autophagy include AMBRA1 (Fimia et al. 2007), UVRAG (Liang et al. 2006) and SH3GLB1 (Takahashi et al. 2007). Binding of BCL2 or BCL2L1 (Bcl-xL) inhibit autophagy (Pattingre et al. 2005, Ciechomska et al. 2009). The inositol 1,4,5-trisphosphate receptor complex that binds BCL2 also interacts with BECN1, inhibiting autophagy (Vincencio et al. 2009). CISD2 (Nutrient-deprivation autophagy factor-1, NAF1), a component in the IP3R complex, interacts with BCL2 at the ER and stabilizes the BCL2-BECN1 interaction (Chang et al. 2010). Starvation leads to activation of c-Jun NH2-terminal kinase-1 (JNK1), which results in the phosphorylation of BCL2 and BCL2L1, which release their binding to BECN1 and thus induces autophagosome formation (Wei et al. 2008). AMBRA1 can simultaneously bind dynein and the Beclin-1 complex. During nutrient starvation, AMBRA1 is phosphorylated in a ULK1-dependent manner (Di Bartolomeo et al. 2010). This phosphorylation releases AMBRA1-associated Beclin-1 complexes from dynein and the microtubule network, freeing the complex to translocate to autophagy initiation sites (Di Bartolomeo et al. 2010). A characteristic of this early phase of autophagosome formation is the formation of PI3P-enriched ER-associated structures called omegasomes (Axe et al. 2008) or cradles (Hayashi-Nishino et al. 2009). Omegasomes appear to concentrate at or near the connected mitochondria-associated ER membrane (Hamasaki et al. 2013). However, the phagophore also can incorporate existing material from other membrane sources such as ER exit sites (ERES), the ER-Golgi intermediate compartment (ERGIC), the Golgi, the plasma membrane and recycling endosomes (Carlsson & Simonsen 2015). Omegasomes lead to the formation of the isolation membrane or phagophore, which is thought to form de novo by an unknown mechanism (Simonsen & Stenmark 2008, Roberts & Ktistakis 2013). Phagophore expansion is probably mediated by membrane uptake from endomembranes and semi-autonomous organelles (Lamb et al. 2013, Shibutani & Yoshimori 2014).ATG9 is a direct target of ULK1. In nutrient-rich conditions mammalian ATG9 is localized to the trans-Golgi network and endosomes (including early, late and recycling endosomes), whereas under starvation conditions it is localized to autophagosomes, in a process that is dependent on ULK1 (Young et al. 2006). ATG9 is believed to play a role in the delivery of vesicles derived from existing membranes to the expanding phagophore (Lamb et al. 2013). Yeast Atg9 forms a complex with Atg2 and Atg18 (Reggiori et al. 2004). PI3P produced at the initiation site is sensed by WIPI2b, the mammalian homologue of Atg18 (Polson et al. 2010). WIPI2b then recruits Atg16L1 (Dooley et al. 2014). There are four WIPI proteins in mammalian cells (Proikas-Cezanne et al. 2015). They are all likely bind PI3P and be recruited to membranes but the function of WIPI1, 3 and 4 in autophagy is not yet clear. WIPI4 (WDR45) has been shown to bind Atg2 and to be involved in lipid droplet formation (Velikkakath et al. 2012); mutations in WIPI4 have been shown to cause a neurodegenerative disease (Saitsu et al. 2013).The elongation of the membrane that will become the autophagosome is regulated by two ubiquitination-like reactions. First, the ubiquitin-like molecule ATG12 is conjugated to ATG5 by ATG7, which acts as an E1-like activating enzyme, and ATG10, which has a role similar to an E2 ubiquitin-conjugating enzyme. The ATG5:ATG12 complex then interacts non-covalently with ATG16L1. This complex associates with the forming autophagosome but dissociates from completed autophagosomes (Geng & Klionski 2008). The second ubiquitin-like reaction involves the conjugation of ubiquitin-like molecules of the LC3 family (Weidberg et al. 2010). LC3 proteins are conjugated through their C-terminal glycine residues with PE by the E1-like ATG7 and E2-like ATG3. This allows LC3 proteins to associate with the autophagosome membrane. The ATG12:ATG5:ATG16L1 complex (Mizushima et al., 2011) acts as an E3 like enzyme for the conjugation of LC3 family proteins (mammalian homologues of yeast Atg8) to phosphatidylethanolamine (PE) (Hanada et al. 2007, Fujita et al. 2008). LC3 PE can be deconjugated by the protease ATG4 (Li et al. 2011, 2012). ATG4 is also responsible for priming LC3 proteins by cleaving the C terminus to expose a glycine residue (Kirisako et al, 2000, Scherz Shouval et al. 2007). LC3 proteins remain associated with autophagosomes until they fuse with lysosomes. The LC3-like proteins inside the resulting autolysosomes are degraded, while those on the cytoplasmic surface are delipidated and recycled. ATG5:ATG12:ATG16L1-positive LC3-negative vesicles represent pre-autophagosomal structures (pre-phagophores and possibly early phagophores), ATG5:ATG12:ATG16L1-positive LC3-positive structures can be considered to be phagophores, and ATG5:ATG12:ATG16L1-negative LC3-positive vesicles can be regarded as mature autophagosomes (Tandia et al. 2011).Phagophore expansion is probably mediated by membrane uptake from endomembranes as well as from semiautonomous organelles (Lamb et al. 2013, Shibutani & Yoshimori 2014).The mechanisms involved in the closure of the phagophore membrane are poorly understood. As the phagophore is a double-membraned structure, its closure involves the fusion of a narrow opening, a process that is distinct from other membrane fusion events (Carlsson & Simonsen 2015). The topology of the phagophore is similar to that of cytokinesis, viral budding or multivesicular body (MVB) formation. These processes rely on the Endosomal Sorting Complex Required for Transport (ESCRT) (Rusten et al. 2012). ESCRT and associated proteins facilitate membrane budding away from the cytosol and subsequent cleavage of the bud neck (Hurley & Hanson 2010). Several studies have shown that depletion of ESCRT subunits or the regulatory ATPase Vps4 causes an accumulation of autophagosomes (Filimonenko et al. 2007, Rusten et al. 2007) but it is not clear whether ESCRTs are required for autophagosome closure or for autophagosome to endosome fusion. UVRAG is also involved in the maturation step, recruiting proteins that bring about membrane fusion such as the class C Vps proteins, which activate Rab7 thereby promoting fusion with late endosomes and lysosomes (Liang et al. 2008)
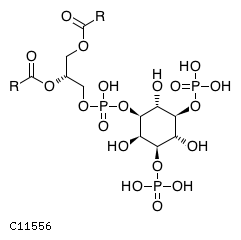
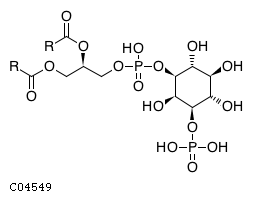
At the plasma membrane, subsequent phosphorylation of phosphatidylinositol 4-phosphate (PI4P) produces phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) and phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3) while the actions of various other kinases and phosphatases produces phosphatidylinositol 3-phosphate (PI3P), phosphatidylinositol 5-phosphate (PI5P), phosphatidylinositol 3,4-bisphosphate (PI(3,4)P2), and phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) (Zhang et al. 1997, Gurung et al. 2003, Guo et al. 1999, Vanhaesebroeck et al. 1997, Tolias et al. 1998, Schaletzky et al. 2003, Kim et al. 2002, Clarke et al. 2010). Many of the phosphatidylinositol phosphatases that act at the plasma membrane belong to the myotubularin family. Enzymatically inactive myotubularin family members can heterodimerize with catalytically active mytotubularins to regulate their stability, activity and/or substrate specificity (Berger et al. 2006, Zou et al. 2012)

 Statistics
Statistics